泛酸激酶及其抑制剂的研究进展
2014-03-08杨延辉赵建新王浩摆茹韩梅
杨延辉,赵建新,王浩,摆茹,韩梅
(宁夏医科大学基础医学院,宁夏 银川 750004)
·综述与专论·
REVIEW AND MONOGRAPH
泛酸激酶及其抑制剂的研究进展
杨延辉,赵建新,王浩,摆茹,韩梅*
(宁夏医科大学基础医学院,宁夏 银川 750004)

辅酶A的生物合成对于病原微生物的生存及致病性至关重要,而作为辅酶A生物合成途径的限速酶——泛酸激酶控制着辅酶A生物合成的起始步骤,且其普遍存在于病原体,并与人体中的同源蛋白相似度极低,故引起人们广泛兴趣,成为潜在的新型抗菌靶标研究的热点,其小分子抑制剂有望开发成为具高选择性、低毒副作用的抗菌药。综述泛酸激酶的分类、特性、晶体结构特征及其调控和成为潜在抗菌药物靶标的可行性,并概述具不同结构类型和抗病原微生物活性的在研泛酸激酶抑制剂。
泛酸激酶;辅酶A;抑制剂;抗病原微生物活性
辅酶A(coenzymeA,CoA,1)广泛存在于生物体内,是转运一碳基团所必需的辅助因子,是体内重要的酰基载体,参与糖代谢、脂肪酸合成与氧化、血红素合成、丙酮酸降解、氨基酸分解和乙酰胆碱合成与乙酰化等代谢过程[1]。CoA生物合成从泛酸(2)开始,需5步酶促反应,分别由coaA、coaB、coaC、coaD和coaE基因编码的酶完成,其间分别生成化合物3~6(见图1)[2],而泛酸激酶(pantothenatekinase,即原核生物中CoaA,真核生物中PanK)又称ATP:D-泛酸-4’-磷酸转移酶(EC.7.1.33),是CoA生物合成途径的限速酶,被认为是抗菌药物开发的潜在新靶点,且其突变与人类神经退行性病变相关[3-4]。鉴于泛酸激酶的重要性,本文对该酶及其抑制剂的研究现状作一综述。
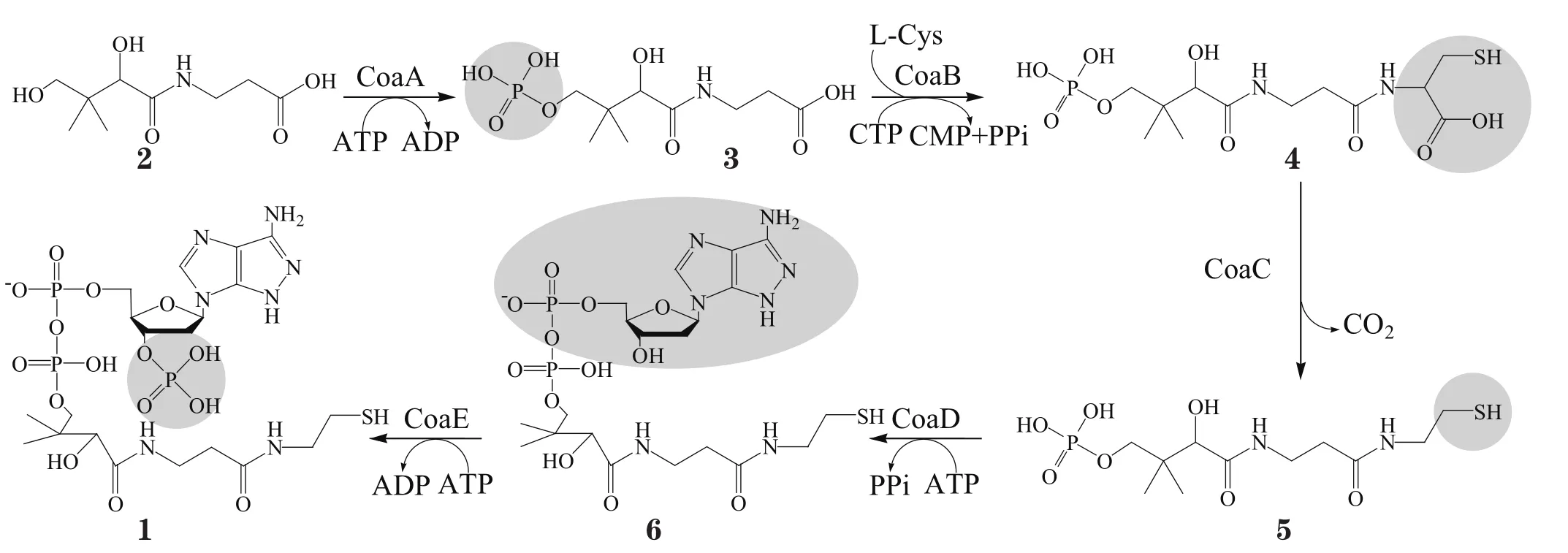
图1 从泛酸开始的CoA生物合成路线Figure 1 Biosynthetic route from pantothenic acid to CoA
1 泛酸激酶的分类
泛酸激酶具有3种类型:Ⅰ型、Ⅱ型和Ⅲ型,其中,Ⅰ型以大肠埃希菌的泛酸激酶(EcCoaA)为代表;Ⅱ型以金葡菌的泛酸激酶(SaCoaA)为代表,主要存在于真核生物中,包括哺乳动物来源的4种亚型:mPanK1、mPanK2、mPanK3和mPanK4;Ⅲ型主要存在于病原微生物中,如幽门螺旋杆菌、铜绿假单胞菌、炭疽芽胞杆菌和结核分枝杆菌(Mtb)等。
在一个生物体中,可以存在多种类型的泛酸激酶,比如Mtb和枯草芽孢杆菌同时含有Ⅰ型和Ⅲ型,但缺乏Ⅱ型[5-6]。迄今,应用比较基因组学进行序列比对时发现,几乎所有生物体(包括古细菌[7])中都含有泛酸激酶相关基因。
2 泛酸激酶的特性
2.1 Ⅰ型泛酸激酶
Ⅰ型泛酸激酶最初是从沙门氏菌和大肠埃希菌中发现。EcCoaA由coaA基因编码,有两个转录起始位点。EcCoaA主要以相对分子质量为36000的同源二聚体形式存在,属于P环激酶超家族,其对泛酸和ATP的亲和常数(Km)分别为36和136μmol·L-1(Song等, J Bacteriol,1992年)。由于结构特点,EcCoaA与底物的结合具有一定的选择特异性[8],酶动力学实验证明其与ATP的结合符合正协同序变模型。Song等(J Bacteriol,1992年)将大肠埃希菌菌株中EcCoaA的表达量提高76倍,却仅使其催化合成的终产物CoA增加了2.7倍,说明EcCoaA可被CoA反馈抑制。另外,CoA硫酯对EcCoaA也有一定的抑制作用(Rock等,J Bacteriol,2003年)。
2.2 Ⅱ型泛酸激酶
Ⅱ型泛酸激酶最初是从构巢曲霉中发现,与Ⅰ型泛酸激酶在序列上无相似性,构巢曲霉的panK基因位于3号染色体上并由3个内含子隔开。Ⅱ型泛酸激酶对泛酸和ATP的Km分别为60和145μmol·L-1,其可被乙酰CoA抑制,而CoA对该酶的反馈抑制作用比CoA硫酯强(Calder等,J Biol Chem,1999年)。
SaCoaA与哺乳动物泛酸激酶(mPanK)的催化中心最为相似,其以同源二聚体形式存在,对泛酸和ATP的Km由 Choudhry等(AntimicrobAgentsChemother,2003年)报道分别为(27±7)和(93±18)μmol·L-1,而Leonardi等[9]报道分别为23和34μmol·L-1,且均表明ATP浓度低时酶活性更好。SaCoaA不会被CoA反馈抑制,其在细胞内催化合成的CoA浓度可累积达毫摩尔级[10]。
mPanK首次在小鼠中发现,其基因由7个内含子和8个外显子组成,而按1号外显子的不同,其又可分成PanK1α和PanK1β两种亚型。PanK1α可被CoA、乙酰CoA和丙二酰CoA抑制,其中CoA抑制作用最弱;PanK1β则不被CoA抑制,乙酰CoA的抑制作用也较弱(Rock等,J Biol Chem,2000年)。
人类的4种PanK的催化中心区域都高度保守,一致性在83%以上。其中,PanK2蛋白靶向于线粒体,并与神经退行性疾病的发生具有一定的相关性[4],而panK2基因位于染色体20p12.3-p12,含有7个外显子,每个外显子上都会发生引起病变的突变,如2号外显子上H173Y[11]和Y227C[12]、3号外显子上D378G[13]与4号外显子上R440P的突变[14]以及5号外显子上可引发PanK2上1142位到1144位的3个核苷酸GAG缺失的突变[15]。人类的泛酸激酶受CoA及其乙酰脂类的抑制[10]。
总之,真核生物中的Ⅱ型泛酸激酶虽然与原核生物的Ⅱ型泛酸激酶具有相似的一级结构,但它们的酶动力学和调控机制等存在显著差异。
2.3 Ⅲ型泛酸激酶
在炭疽芽胞杆菌中发现,泛酸激酶基因coaX能修复大肠埃希菌coaA15(Ts)缺陷株,表明该基因编码一种新型泛酸激酶,并命名为CoaX[16],该酶属于ASKHA家族。炭疽芽胞杆菌和幽门螺旋杆菌中的泛酸激酶对泛酸的Km分别为168和101μmol·L-1,对ATP的Km分别为3和10mmol·L-1[17]。Ⅲ型泛酸激酶不受CoA和乙酰CoA的抑制,且不以N取代的泛酸酰胺类药物为底物催化合成类似于CoA的抗代谢物[18]。
综上所述,3种类型的泛酸激酶在氨基酸序列、调控模式以及对底物的亲和性和特异性上都有很大差异,表1对它们的特性进行了汇总比较并列举了其代表性抑制剂。
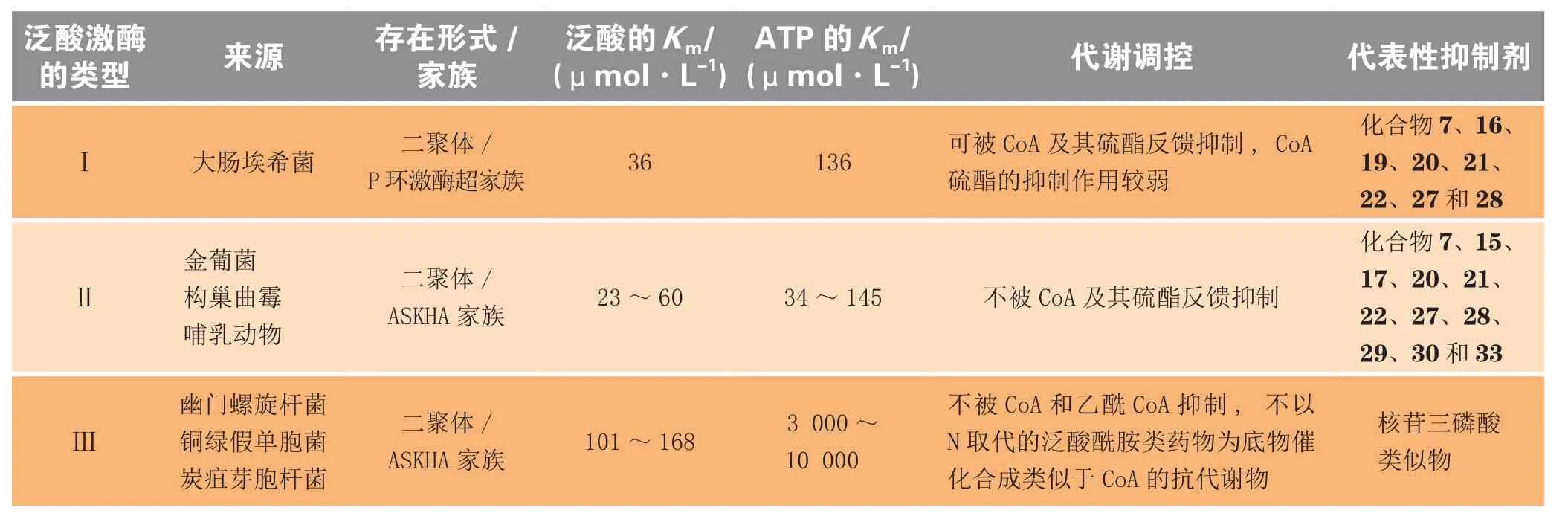
表1 3种类型泛酸激酶的特性比较及其代表性抑制剂Table 1 Comparison of the properties of three types of pantothenate kinases and their representative inhibitors
3 泛酸激酶晶体结构特征
Ⅰ型泛酸激酶与CoA或ATP类似物AMPPNP的复合物晶体结构显示,CoA的α和β磷酸与AMPPNP的β和γ磷酸占据了激酶的相同位点Lys101。而CoaA·ADP·泛酸三元复合物的结构分析明确了CoaA上与泛酸结合的氨基酸残基,且经序列比对发现,这些氨基酸残基在多种细菌中都是保守的,尤其是Asp127和Tyr240[19]。经含有CoaA·ADP·泛酸和CoaA·CoA的泛酸激酶晶体结构叠合对比,发现泛酸、ADP和CoA使用了相同的结合口袋;而将泛酸酰胺类似物N5-Pan和泛酸激酶进行分子对接,则发现每种配体的结合都会产生一种不同的蛋白构象[20]。
Ⅱ型和Ⅲ型泛酸激酶都含有核糖核酸酶H类似的结构。SaCoaA的泛酸结合口袋可与ATP牢固结合,需要一价阳离子来抵消口袋的电荷,从而削弱口袋对ATP的结合,使ATP易于与口袋解离,而活性中心人PanK2与SaCoaA的结构和底物结合机制均相似。然而,泛酸结合到铜绿假单胞菌的Ⅲ型泛酸激酶催化口袋后,酶活性中心的P环结构可与ATP的腺嘌呤部分特异性匹配,但该酶与底物形成的氢键和范德华力弱于Ⅱ型激酶,导致其与ATP的结合能力不及Ⅱ型激酶[18]。而且,由于Ⅲ型泛酸激酶二聚体的活性中心口袋相对狭小,使得泛酸及其类似物(如泛酸酰胺类化合物)不易进入酶活性中心。所以,含Ⅲ型泛酸激酶的细菌会对泛酸酰胺类抗生素产生耐药性。
4 辅酶A生物合成和泛酸激酶的调控
CoA的生物合成调节方式主要包括体内CoA的区域化分布、合成过程的反馈调节、CoA合成的关键酶的基因表达调节和CoA的降解调节[10]。在CoA的生物合成途径中,最主要的调控酶是泛酸激酶和4-磷酸泛酰巯基乙胺腺苷转移酶(CoaD)。CoA可与ATP酶的活性中心竞争性结合,从而对其生物合成产生负调控作用(Rock等,J Biol Chem,2000年),而CoaD则主要由CoA与底物4’-磷酸泛酰巯基乙胺竞争性结合于酶活性中心位点而被反馈抑制。泛酸激酶的基因表达水平较低有2个原因:1)coaA基因的启动子与其他基因的启动子同源性低;2)泛酸激酶使用了稀有密码子[10]。
5 泛酸激酶成为潜在抗菌药物靶标的可行性
几乎所有病原微生物中都存在CoaA基因,且有些病原微生物中CoaA基因过表达或者含有多拷贝coaA基因,如炭疽芽孢杆菌和Mtb等。
真核和原核生物的泛酸激酶研究成果表明,它们的序列同源性极低,即使有些在序列上具有相似性,但其动力学特性和调节方式仍存在差异,且泛酸激酶的天然抑制剂CoA及其硫酯对不同来源的泛酸激酶具有不同的抑制活性。此外,Mtb与大肠埃希菌中的Ⅰ型泛酸激酶(MtbCoaA和EcCoaA)在结构上非常相似(见图2),且泛酸结合区域的氨基酸序列非常保守,但在进行酶促反应时其配基结合位点会发生改变(见图3)。再者,MtbCoaA对泛酸和ATP的Km都高于EcCoaA。因此,不同来源的泛酸激酶的结构和活性特异性使得设计与开发出专门针对于病原微生物,且选择特异性高的CoaA抑制剂成为可能[21-22],CoaA也就成为抑制CoA生物合成和病原微生物生长的首选靶标。
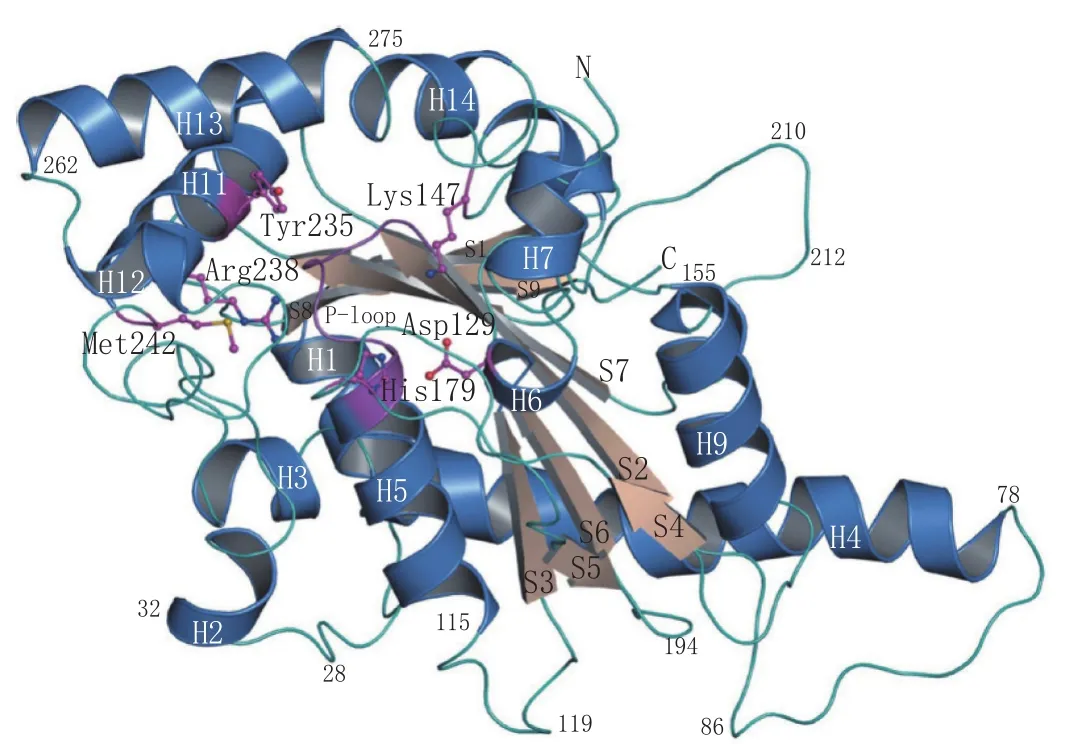
图2 MtbCoaA的结构Figure 2 Structure of MtbCoaA
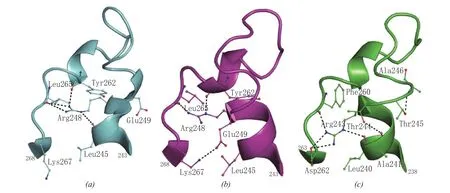
图3 泛酸激酶中泛酸结合区域的构象Figure 3 Conformation of the pantothenate-binding region in pantothenate kinase
6 泛酸激酶抑制剂
截至目前发现的CoaA抑制剂化合物种类主要包括 泛磺酸及其衍生物、泛醇、N-泛酰取代胺、泛酰肼及其衍生物、泛氨酮(pantothenone)及其衍生物、泛酸类似物、N-取代泛酰胺(pantothenamide)、苯三唑类化合物、联芳乙酸衍生物和天然泛酸类似物等(见图4)。
6.1 泛磺酸及其衍生物
泛磺酸(7)是首个报道具有体外抗菌活性的泛酸类似物,对大肠埃希菌、乳酸菌、链球菌和白喉棒状杆菌等都有抑制活性,其体外MIC分别为4.8~6.1g·L-1、3.7~13.8g·L-1、0.9~4.0g·L-1和2~50μmol·L-1,且它对不同细菌的抑制活性同介质中泛酸的浓度有很大关系,即至少泛磺酸的浓度较泛酸高500倍以上,其才能发挥抗菌作用(McIlwain,Biochem J,1942年)。体内研究表明,以2.2g·kg-1剂量给予泛磺酸,对高致病性链球菌感染的大鼠能起保护作用(McIlwain等,Lancet,1943年)。需要如此高的治疗剂量方能显效,是限制泛磺酸进一步研发的主要因素。
泛磺酰胺(8)是泛酸的磺胺类似物,溶解性低于泛磺酸,使其在血液中维持时间更长,其以2.2g·kg-1剂量给予模型大鼠所产生的活性优于同剂量泛磺酸。泛磺酸衍生物9、10和11a体外抗菌的MIC同泛磺酸接近,而衍生物11b和11c的体外抗菌活性则有所提高,且11b具有比泛磺酸更好的体内活性,但衍生物9的毒性较高。合成的芳基取代泛磺酸衍生物12和13无体外抗菌活性,N-取代泛磺酰胺(14a-d)的体外抗链球菌活性优于泛磺酸。而且,泛磺酸及其衍生物仅有右旋结构具抑菌活性(White等,J Am Chem Soc,1946年;Winterbottom等,J Am Chem Soc,1947年)。
6.2 泛醇和N-泛酰取代胺
曾有研究认为泛醇(15)能占据泛酸激酶的活性中心并产生竞争性抑制作用,而Kumar等[23]的研究发现泛酸可致使泛醇磷酸化。泛醇对含Ⅱ型泛酸激酶的金葡菌具有抑制作用,对泛酸激酶的IC50可达64μmol·L-1,对该菌的MIC为2mmol·L-1[24]。而ω-甲基泛醇(16)对乳酸菌的MIC约为100μmol·L-1(Drell等,Arch Biochem Biophys,1954年)。不过,右旋泛醇抗肠膜状明串珠菌(Leuconotoc mesenteroides)的活性优于泛磺酸,但抗其他微生物的作用不及泛磺酸,且在体内会转化为泛酸(Snell等,J Biol Chem,1945年),因此它不是非常理想的抗菌剂。
为解决上述问题,人们合成了一系列泛醇衍生物——N-泛酰取代胺(17)。其中,N-泛酰烷基胺(17c)抗乳酸菌的活性最好,其次是化合物17a,它在浓度高于泛酸5倍时即能有效抑制乳酸菌的生长,而化合物17b再次之,但其活性仍优于泛醇;化合物17d的抗链球菌活性最好;化合物17e也具有体外抗菌和抗疟原虫活性[25],且在体内不会转化成泛酸,其体内活性有待进一步研究。
6.3 泛酰肼及其衍生物
Madinaveitia等合成了泛酰肼及其衍生物(18~21),并在体外实验中发现,在10μmol·L-1的D,L-泛酸存在的情况下,泛酰肼(18)抑制乳酸菌的活性略强于泛磺酸,且将D,L-泛酸浓度再提高0.1μmol·L-1时,其抗菌活性可提高50倍以上;泛酰胺(19)、化合物20和21都具有抑制乳酸菌活性,而化合物20和21还具有抑制链球菌活性(Madinaveitia等,BiochemJ,1945年)。这类化合物的抗菌谱及体内抗菌活性尚有待进一步研究。
6.4 泛氨酮及其衍生物
D-甲基泛氨酮(22)和D-苯基泛氨酮(23)均属于非泛酸竞争性抑制剂,前者对乳酸菌和酵母菌的IC50分别为460和2300μmol·L-1,后者对干酪乳杆菌(Lactobacillus casei)、溶血性链球菌、大肠埃希菌、金葡菌、产脂内孢霉(Endomyces vernalis)和酿酒酵母菌(Saccharomyces cerevisiae)的IC50分别为190、220、7200、500、140和120μmol·L-1。此外,D-对甲苯基泛氨酮(24)也具有抗乳酸菌活性[25]。
6.5 泛酸类似物
ω-甲基泛酸(25)在体内外实验中都能显示出抗溶血性链球菌活性,其MIC约为50μmol·L-1,但它也能引起哺乳动物体内泛酸缺乏;此外,β取代的泛酸类似物的抗菌活性强于ω取代类似物(Drell等,Arch Biochem Biophys,1954年)。高泛酰牛磺酸(26)是泛酸激酶的竞争性抑制剂,但其抗菌作用不及泛磺酸(Mcilwain等,Biochem J,1942年)。化合物(27)为I型泛酸激酶的抑制剂[26],目前尚无抗菌活性数据。
6.6 N-取代泛酰胺
N-取代泛酰胺衍生物发挥抗菌活性的作用机制是,通过竞争性结合于泛酸激酶而干扰CoA的代谢[27]。N-戊基泛酰胺(28a)和N-庚基泛酰胺(28b)具有抗金葡菌活性,MIC分别为25和0.16μmol·L-1,它们虽然对I型和Ⅱ型泛酸激酶有抑制活性,却对Ⅲ型酶无抑制作用[28]。这类泛酸激酶抑制剂可通过体内代谢而转化为可使酰基载体蛋白失活的化合物,从而进一步有效抑制细菌细胞膜的生物合成。体外抗菌活性研究发现,末端具有反式烯丙基的N-戊基泛酰胺(29)的抗菌活性较好,对敏感和耐药金葡菌的MIC可达3.2μmol·L-1,对大肠埃希菌的MIC为2μmol·L-1(Strauss等,J Biol Chem,2002年)。最新研究表明,N-戊基泛酰胺末端二烃基构象的改变能使其显示出不同的生物学活性,当末端的反式烯丙基变为顺式(30)时,其具有抗疟原虫活性,IC50为(2.4±0.2)μmol·L-1[29]。
6.7 苯三唑和联芳乙酸衍生物
针对Mtb的Ⅰ型泛酸激酶开展的研究发现,苯三唑类化合物(31)具有泛酸激酶抑制活性,IC50可达0.05μmol·L-1,可遗憾的是其无体外抗Mtb活性。因此,需对此类化合物开展进一步的构效关系研究,以发现具有抗菌活性的泛酸激酶抑制剂[21-30]。另有研究发现,联芳乙酸衍生物(32a-c)具有体外抗Mtb作用,MIC为4~16mg·L-1[22]。
6.8 天然泛酸类似物
CJ-15801(33)是从真菌Seimatosporium sp.CL28611中分离得到的1个天然泛酸类似物,同泛酸的差异是β-丙氨酸部分含有一个双键,其对多药耐药金葡菌具有抑制作用,MIC为6.25~50mg·L-1[31]。目前,CJ-15801及其酯类衍生物已被全合成[32-33],为进一步开展此类化合物的抗菌谱及体内外活性研究奠定了物质基础。
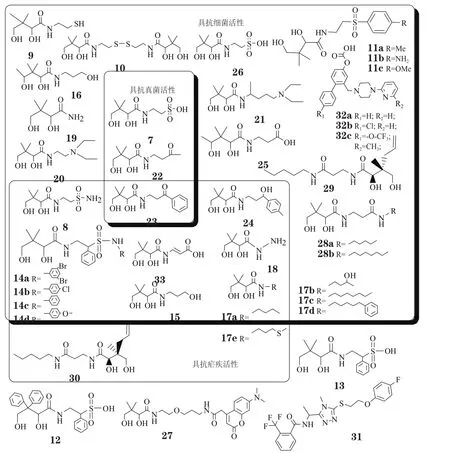
图4 截至目前发现的泛酸激酶抑制剂结构类型Figure 4 The structure types of pantothenate kinase inhibitors found so far
7 结语
鉴于世界范围内多药和极端耐药病原菌不断增多,急需开发新的药物靶点和治疗策略,而探索并鉴定可能成为新型抗菌药物靶点的病原微生物关键酶,则是意义深远的创新性研究。综上所述,CoA在生物体内的诸多代谢过程中发挥重要作用,而泛酸激酶是CoA生物合成中的限速酶,且病原微生物与哺乳动物中的泛酸激酶在序列上、代谢动力学和调节上均存在显著差异,使得泛酸激酶成为新型抗菌药物研发的理想靶点,并有可能设计和开发出特异性靶向病原菌的泛酸激酶抑制剂。目前泛酸激酶抑制剂设计的主要思路包括寻找底物类似物和反应过渡态类似物以及建立高通量筛选模型对已有的化合物库进行筛选,藉此已发现了一些泛酸激酶抑制剂,其中CoA类似物是最具特异性抑制活性的化合物,有些还表现出体内外抗菌活性。当然,眼下的相关研究仍处于探索阶段,以泛酸激酶作为有效靶标开发出高选择性的抗病原微生物药物,尚需大量的实验研究支持。
[1]Horvath Z, Vecsei L. Current medical aspects of pantethine[J]. Ideggyogy Sz, 2009, 62(7/8): 220-229.
[2]Ishibashi T, Tomita H, Yokooji Y, et al. A detailed biochemical characterization of phosphopantothenate synthetase, a novel enzyme involved in coenzyme A biosynthesis in the Archaea[J]. Extremophiles, 2012, 16(6): 819-828.
[3]Ma L Y, Wang L, Yang Y M et al. Novel gene mutations and clinical features in patients with pantothenate kinase-associated neurodegeneration[J]. Clin Genet, 2014. doi: 10.1111/cge.12341. [Epub ahead of print].
[4]Tanteles G A, Spanou-Aristidou E, Antoniou C, et al. Novel homozygous PANK2 mutation causing atypical pantothenate kinaseassociated neurodegeneration (PKAN) in a Cypriot family[J]. J Neurol Sci, 2014, 340(1/2): 233-236.
[5]Awasthy D, Ambady A, Bhat J, et al. Essentiality and functional analysis of type I and type III pantothenate kinases of Mycobacterium tuberculosis[J]. Microbiology, 2010, 156(Pt 9): 2691-2701.
[6]Ogata Y, Katoh H, Asayama M, et al. Role of prokaryotic type I and III pantothenate kinases in the coenzyme A biosynthetic pathway of Bacillus subtilis[J]. Can J Microbiol, 2014, 60(5): 297-305.
[7]Tomita H, Yokooji Y, Ishibashi T, et al. Biochemical characterization of pantoate kinase, a novel enzyme necessary for coenzyme A biosynthesis in the Archaea[J]. J Bacteriol, 2012, 194(19): 5434-5443.
[8]Awuah E, Ma E, Hoegl A, et al. Exploring structural motifs necessary for substrate binding in the active site of Escherichia coli pantothenate kinase[J] . Bioorg Med Chem, 2014, 22(12): 3083-3090.
[9]Leonardi R, Chohnan S, Zhang Y M, et al. A pantothenate kinase from Staphylococcus aureus refractory to feedback regulation by coenzyme A[J]. J Biol Chem, 2005, 280(5): 3314-3322.
[10]Leonardi R, Zhang Y M, Rock C O, et al. Coenzyme A: back in action[J]. Prog Lipid Res, 2005, 44(2/3): 125-153.
[11]Saleheen D, Nazir A, Khanum S, et al. A novel mutation in a patient with pantothenate kinase-associated neurodegeneration[J]. CMAJ, 2005, 173(6): 578-579.
[12]Saleheen D, Ali T, Aly Z, et al. Novel mutation in the PANK2 gene leads to pantothenate kinase-associated neurodegeneration in a Pakistani family[J]. Pediatr Neurol, 2007, 37(4): 296-298.
[13]Kim J, Shin H, Youn J, et al. A novel PANK2 gene mutation with sudden-onset dystonia[J]. Can J Neurol Sci, 2012, 39(3): 395-397.
[14]Seo J H, Song S K, Lee P H. A novel PANK2 mutation in a patient with atypical pantothenate-kinase-associated neurodegeneration presenting with adult-onset parkinsonism[J]. J Clin Neurol, 2009, 5(4): 192-194.
[15]Rump P, Lemmink H H, Verschuuren-Bemelmans C C, et al. A novel 3-bp deletion in the PANK2 gene of Dutch patients with pantothenate kinase-associated neurodegeneration: evidence for a founder effect[J]. Neurogenetics, 2005, 6(4): 201-207.
[16]Paige C, Reid S D, Hanna P C, et al. The type III pantothenate kinase encoded by coaX is essential for growth of Bacillus anthracis[J]. J Bacteriol, 2008, 190(18): 6271-6275.
[17]Brand L A, Strauss E. Characterization of a new pantothenate kinaseisoform from Helicobacter pylori[J]. J Biol Chem, 2005, 280(21): 20185-20188.
[18]Yang K, Strauss E, Huerta C, et al. Structural basis for substrate binding and the catalytic mechanism of type III pantothenate kinase[J]. Biochemistry, 2008, 47(5): 1369-1380.
[19]Ivey R A, Zhang Y M, Virga K G, et al. The structure of the pantothenate kinase.ADP.pantothenate ternary complex reveals the relationship between the binding sites for substrate, allosteric regulator, and antimetabolites[J]. J Biol Chem, 2004, 279(34): 35622-35629.
[20]Hughes S J, Antoshchenko T, Kim K P, et al. Structural characterization of a new N-substituted pantothenamide bound to pantothenate kinases from Klebsiella pneumoniae and Staphylococcus aureus[J]. Proteins, 2014, 82(7): 1542-1548.
[21]Bjorkelid C, Bergfors T, Raichurkar A K, et al. Structural and biochemical characterization of compounds inhibiting Mycobacterium tuberculosis pantothenate kinase[J]. J Biol Chem, 2013, 288(25): 18260-18270.
[22]Venkatraman J, Bhat J, Solapure S M, et al.Screening,identifcation,and characterization of mechanistically diverse inhibitors of the Mycobacterium tuberculosis enzyme, pantothenate kinase (CoaA)[J]. J Biomol Screen, 2012, 17(3): 293-302.
[23]Kumar P, Chhibber M, Surolia A. How pantothenol intervenes in Coenzyme-A biosynthesis of Mycobacterium tuberculosis[J]. Biochem Biophys Res Commun, 2007, 361(4): 903-909.
[24]Chohnan S, Murase M, Kurikawa K, et al. Antimicrobial activity of pantothenol against staphylococci possessing a prokaryotic type II pantothenate kinase[J]. Microbes Environ, 2014, 29(2): 224-226.
[25]Spry C, Kirk K, Saliba K J. Coenzyme A biosynthesis: an antimicrobial drug target[J]. FEMS Microbiol Rev, 2008, 32(1): 56-106.
[26]Meier J L, Mercer A C, Rivera H, et al. Synthesis and evaluation of bioorthogonalpantetheineanaloguesforinvivoproteinmodifcation[J].J Am Chem Soc, 2006, 128(37): 12174-12184.
[27]Spry C, Macuamule C, Lin Z, et al. Pantothenamides are potent, ontarget inhibitors of Plasmodium falciparum growth when serum pantetheinase is inactivated[J]. PLoS One, 2013, 8(2): e54974.
[28]Thomas J, Cronan J E. Antibacterial activity of N-pentylpantothenamide is due to inhibition of coenzyme a synthesis[J]. Antimicrob Agents Chemother, 2010, 54(3): 1374-1377.
[29]Hoegl A, Darabi H, Tran E, et al. Stereochemical modification of geminal dialkyl substituents on pantothenamides alters antimicrobial activity[J]. Bioorg Med Chem Lett, 2014, 24(15): 3274-3277.
[30]Reddy B K, Landge S, Ravishankar S, et al. Assessment of Mycobacterium tuberculosis pantothenate kinase vulnerability through target knockdown and mechanistically diverse inhibitors[J]. Antimicrob Agents Chemother, 2014, 58(6): 3312-3326.
[31]Van der Westhuyzen R, Hammons J C, Meier J L, et al. The antibiotic CJ-15,801 is an antimetabolite that hijacks and then inhibits CoA biosynthesis[J]. Chem Biol, 2012, 19(5): 559-571.
[32]Gunanathan C, Ben-David Y, Milstein D. Direct synthesis of amides from alcohols and amines with liberation of H2[J]. Science, 2007, 317(5839): 790-792.
[33]Kim J, Moon Y, Lee S, et al. A Pd-catalyzed one-pot dehydrogenative aromatization and ortho-functionalization sequence of N-acetyl enamides[J]. Chem Commun (Camb), 2014, 50(24): 3227-3230.
Pantothenate Kinase and Its Inhibitors:Current Research Status
YANG Yanhui, ZHAO Jianxin, WANG Hao, BAI Ru, HAN Mei
( School of Basic Medicine, Ningxia Medical University, Yinchuan 750004, China)
The biosynthesis of coenzyme A(CoA) plays a vital role in the survival and virulence of pathogenic microorganism. Pantothenate kinase as the rate-limiting enzyme controls the starting step in CoA biosynthesis, which is widespread in the pathogen and of which the similarity to homologous proteins in human is extremely low. Therefore, the kinase has gained much of people’s interest and has become a hot spot of research for potential new type of antibacterial targets. Its small molecule inhibitors are expected to be developed into the antimicrobials with high selectivity and lowtoxicity.Theclassifcation,propertyandcrystalstructureofpantothenatekinaseanditsregulationaswellasthefeasibilityofitsuseasapotentialantibacterial target were reviewed. The pantothenic acid kinase inhibitors in development with different structures and activities against pathogenic microorganism were summarized.
pantothenate kinase; coenzyme A; inhibitor; anti- pathogenic microorganism activity
Q936; R978
A
1001-5094(2014)09-0641-08
接受日期:2014-08-27
项目资助:宁夏医科大学特殊人才科研启动项目(No.XT201319)
*通讯作者:韩梅,教授;
研究方向:病原生物学与免疫学;
Tel:0951-6980122;E-mail:hanmei0708@126.com
