缺氧预处理减轻内质网应激所致的大鼠心肌微血管内皮细胞损伤*
2013-10-24武旭东张振英王晓礽李玉珍刘秀华
武旭东, 张振英, 王晓礽, 李玉珍, 刘秀华△
(中国人民解放军总医院 1门诊部, 2病理生理研究室,北京 100853)
·论著·
缺氧预处理减轻内质网应激所致的大鼠心肌微血管内皮细胞损伤*
武旭东1△▲, 张振英2▲, 王晓礽2, 李玉珍2, 刘秀华2△
(中国人民解放军总医院1门诊部,2病理生理研究室,北京 100853)
目的研究缺氧预处理(hypoxic preconditioning, HPC)对内质网应激(endoplasmic reticulum stress, ERS)所致的心肌微血管内皮细胞(microvascular endothelial cells, MVECs)损伤的影响。方法以毒胡萝卜素(thapsigargin, TG)诱导大鼠心肌MVECs ERS,以乳酸脱氢酶(lactate dehydrogenase, LDH)漏出和细胞凋亡率检测细胞损伤, phalloidin-FITC荧光染色和内质网染色分别观察细胞骨架和细胞内质网形态变化,双向电泳-质谱技术检测TG作用后内皮细胞蛋白质谱表达变化,Western blotting技术检测ERS相关的分子表达。结果TG剂量依赖性诱导MVECs LDH漏出和细胞凋亡,并出现内质网应激分子钙网蛋白(calreticulin, CRT)和葡萄糖调节蛋白78(glucose-regulated protein 78, GRP78)表达上调;HPC减轻TG诱导的MVECs损伤,并可以抑制TG诱导的ERS相关分子的表达上调。结论HPC减轻内质网应激所致的大鼠心肌MVECs损伤。
缺氧预处理; 微血管内皮细胞; 内质网应激
高血压、动脉粥样硬化、冠心病等心血管疾病严重危及人类健康,血管内皮功能障碍(endothelial dysfunction)是上述疾病的共同发病环节,决定疾病的发生、发展和疗效,改善内皮功能已成为心血管疾病治疗的新靶点[1],探索激发机体内源性保护机制,增强内皮细胞对缺血缺氧的抵抗能力,是减轻内皮功能障碍的重要策略。缺血预处理是指预先短暂缺血可以延缓或减轻组织后续缺血/再灌注损伤的现象,可以被缺氧预处理(hypoxic preconditioning, HPC)所模拟[2],是迄今已知最强的内源性保护现象之一,其机制涉及减轻细胞内钙超载、减轻氧化应激、诱导内源性保护蛋白合成等多种作用。内质网 (endoplasmic reticulum, ER) 是细胞内膜/分泌蛋白合成的细胞器,在调节蛋白质正确折叠、钙稳态和细胞凋亡等方面具有重要作用。血管内皮细胞具有高度发达的内质网,对应激刺激非常敏感,缺血、缺氧等多种因素均可以诱导内质网未折叠/错误折叠蛋白质的聚集和钙稳态破坏等功能紊乱状态,即内质网应激(endoplasmic reticulum stress, ERS)。毒胡萝卜素(thapsigargin, TG)通过抑制内质网钙-ATPase、排空内质网钙离子诱导细胞严重内质网应激反应。长期或过度内质网应激触发C/EBP同源蛋白[CCAAT/enhancer-binding protein (C/EBP) homologous protein, CHOP]、caspase-12等内质网相关细胞死亡途径,是缺血再灌注损伤、动脉粥样硬化、糖尿病等[3]疾病的重要发病机制。CHOP是含有169(人)或168(啮齿类)个氨基酸残基的分子量为29 kD的蛋白,内质网应激时CHOP在转录水平上被诱导,其过表达可导致生长停滞和细胞凋亡。研究证实HPC通过调动内源性保护机制减轻钙超载、氧化应激等病理过程而发挥细胞保护作用[4],但是HPC是否可以直接抑制过度ERS、减轻CHOP介导的凋亡而发挥细胞保护作用尚不清楚。本实验在原代培养大鼠心肌微血管内皮细胞(microvascular endothelial cells, MVECs)模型上,采用ERS诱导剂TG诱导ERS,研究HPC对ERS 致MVECs损伤的影响及其机制。
材 料 和 方 法
1主要试剂
M199培养基为Gibco产品;内皮细胞生长支持物(endothelial cell growth supplement, ECGS)、Ⅰ型胶原酶、FITC-albumin、phalloidin-FITC、乙二胺四乙酸(ethylenediaminetetraacetic acid, EDTA)、羟乙基哌嗪乙磺酸(hydroxyethyl piperazine ethanesulfonic acid, HEPES)、苯甲基磺酰氟(phenylmethylsulfonyl fluoride, PMSF)、Triton X-100、十二烷基硫酸钠(sodium dodecyl sulfate, SDS)、钒酸钠(Na2VO4)、丙烯酰胺、甲叉丙烯酰胺、二硫苏糖醇(dithiothreitol, DTT)、leupeptin、四甲基乙二胺(tetramethylethylenediamine, TEMED)和过硫酸胺(ammonium persulfate, APS)均为Sigma产品;蛋白电泳分子量(7~175 kD)标记为Bio-Rad产品;总RNA提取试剂盒(DP419)、cDNA第1链合成试剂盒(KR104)、2× Taq PCR MasterMix(KT201)、100 bp DNA Ladder (MD109)和电泳级琼脂糖(RT101)均购自北京天根生化科技有限公司;兔抗人钙网蛋白(calreticulin, CRT; SPA-600)和兔抗大鼠葡萄糖调节蛋白78(glucose-regulated protein 78, GRP78; SPA-826)多克隆抗体购自Stressgen;兔抗人磷酸甘油醛脱氢酶(glyceraldehydes-3-phosphate dehydrogenase, GAPDH; Sc-25778)、兔抗小鼠CHOP(Sc-575)、兔抗人Bax(Sc-493)和兔抗人Bcl-2(Sc-783)多克隆抗体和增强化学发光(enhanced chemiluminescence, ECL;Sc-2048)试剂盒均购自Santa Cruz;辣根过氧化酶标记山羊抗兔(80259)和山羊抗小鼠IgG(79659)购自Jackson ImmunoResearch;TG(586005)购自Israel Calbiochem;乳酸脱氢酶(lactate dehydrogenase, LDH)测定试剂盒为南京建成生物工程研究所产品;Annexin V-FITC细胞凋亡检测试剂盒购自南京凯基生物工程公司;内质网形态染色试剂盒购自上海杰美基因医药科技有限公司。固相pH梯度(immobilized pH gradient, IPG) buffer (pH 3~10)、 Destreak Reagent、低分子量marker、上样滤纸和IPG干胶条(pH 3~10)为Amersham Biosciences产品;蛋白酶抑制剂(cocktail tablets)为Roche产品;碘乙酰胺为Acros organics产品;乙腈(色谱级)为J. T. Baker产品;三氟乙酸(trifluoracetic acid,TFA)为Merck产品;α-氰基-4-羟基肉桂酸(α-cyano-4-hydroxycinnamic acid,CHCA;质谱基质)购自Bruker。其余化学试剂为国产分析纯产品。
2心肌微血管内皮细胞原代培养及分组
根据本室建立的大鼠MVECs培养方法进行培养[5]。大鼠颈椎脱臼快速处死后,无菌操作取出大鼠心脏,去除心房、右心室以及心内膜和心外膜等组织后;剪碎心肌组织,0.1% I型胶原酶振荡消化10 min,继而0.1%胰蛋白酶消化10 min,重复消化1次后收集消化液,用100 μm滤网过滤,收集滤液,室温下1 500 r/min离心10 min收集血管段,用M199培养液(新生牛血清20%,ECGS 0.02 g/L,谷氨酰胺2 mmol/L,青霉素1×105U/L,链霉素100 mg/L)悬浮血管段,于37 ℃、5%CO2培养箱中孵育4 h后换液,以后每3 d换培养液1次直至细胞单层融合后进行传代培养。以内皮细胞特异性表达的黏附分子和在体内皮细胞完整性标志分子VE-cadherin 抗体(Santa Cruz) 进行细胞免疫荧光染色,鉴定培养的心肌MVECs,其纯度为90%以上。细胞按1×104/cm2传代用于细胞骨架染色和内质网形态测定;按5×104/cm2进行传代用于其它各项测定。同步化处理24 h后,随机分为下列各组:(1) 正常对照组(control):细胞常规培养至实验结束;(2) 内质网应激组(TG组):向培养液内加入TG(终浓度分别为0.1、0.2、0.5、1 μmol/L),置于细胞培养箱内常规培养24 h结束实验;(3) HPC组:按照本室报道的方法[6]将细胞置于缺氧仓内,通入95% N2-5% CO2混合气20 min后,常规培养24 h结束试验;(4) HPC+TG组:按照步骤(3)进行HPC后,向培养液内加入0.5 μmol/L TG 孵育24 h结束实验。
3培养基LDH活性和细胞凋亡率检测
实验结束后收集各组细胞培养基,参照LDH试剂盒操作说明进行LDH活性测定。0.2%胰蛋白酶收集MVECs后,按照试剂盒说明分别加入Annexin V和PI,室温孵育15 min后,流式细胞仪(Becton-Dickinson)检测细胞凋亡率变化。
4细胞通透性检测
MVECs按1×105/cm2接种于铺有1%明胶的双层通透的培养皿(Transwell)顶层小室微孔膜上(直径6.5 mm,孔径为3 mm),待细胞长至融合,按上述实验分组处理细胞后,参照Gillrie等[7]报道方法进行通透性测定:每个双层小室的顶室加入100 μL (终浓度250 mg/L) FITC-albumin, 37 ℃孵育4 h后,分别取顶室培养液20 μL和底室培养液100 μL于96孔板内,荧光酶标仪测定其荧光强度。内皮细胞单层对白蛋白的通透性采用通透率表示:通透率(%)=底室荧光强度/(底室荧光强度+顶室荧光强度),底室和顶室荧光强度分别采用相应体积校正。
5细胞骨架染色
参照Zhang等[8]报道的方法,MVECs种于激光共聚焦专用培养皿(Mattek),实验结束后PBS洗涤2次,2.5%戊二醛固定30 min,1%牛血清白蛋白封闭30 min后,加入phalloidin-FITC(终浓度5 mg/L)室温孵育1 h,PBS洗涤3次后加入DAPI标记的封片剂,在激光扫描共聚焦显微镜(FV1000, Olympus)下观察并采集图像。
6双向电泳-质谱分析
MVECs总蛋白质提取及双向电泳-质谱分析按照本室以往报道方法进行[5], Bradford法蛋白定量后分装、-70 ℃保存。参照改进的胶内加样方法,上样量为1 mg蛋白,平衡后的一向电泳后的IPG胶条置于二向电泳仪(Amersham Pharmacia)上进行SDS-PAGE。凝胶常规固定、考马斯亮蓝染色。图像扫描后利用Image Master 2D Elite图像分析(Amersham Pharmacia)软件进行分析。选取各组凝胶中Volume值[即蛋白点的面积(area)与灰度值(intensity)的积分]相差≥2.0倍的蛋白斑点,胶内酶切后进行质谱分析,经Bruker Daltonics Bio Tools 2.2软件处理后,采用Mascot软件在NCBI nr数据库中进行检索。
7RT-PCR
按照试剂盒操作说明提取培养内皮细胞总RNA,两步法行RT-PCR,总RNA逆转录参照cDNA第1链合成试剂盒操作说明,各目的基因上、下游引物(由北京三博远志生物技术公司合成)见表1,PCR扩增反应结束后取5 μL PCR产物进行1.5% 琼脂糖凝胶电泳,电泳图像采用Image-Pro Plus 图像分析软件(Version 4.1)分析平均吸光度值,以目的片段和GAPDH的平均吸光度比值反映目的基因mRNA的相对水平。

表1 PCR引物序列
GRP78: glucose-regulated protein 78; CRT: calreticulin; CHOP: C/EBP homologous protein.
8Westernblotting检测
按本室以往报道的方法[5]提取MVECs总蛋白,Bradford法蛋白定量后分装、-70 ℃保存。取上述蛋白提取液上清(含蛋白60 μg)进行SDS-PAGE(10%分离胶),将电泳分离后的蛋白质电转移至硝酸纤维素膜上,经10%脱脂奶粉封闭后分别加入CRT(1∶1 000)、GRP78(1∶1 000)、CHOP(1∶200)、Bax(1∶200)、Bcl-2(1∶200)、GAPDH(1∶500)抗体,室温孵育4 h,洗膜后以相应的Ⅱ抗室温孵育1 h。ECL法显影后暗室X光胶片曝光,采用Image-Pro Plus 图像分析软件(Version 4.1)分析蛋白条带的积分吸光度值(integral absorbance,IA),IA=平均吸光度值×面积,以目的蛋白IA值/GAPDHIA值的比值反映目的蛋白相对水平。
9内质网形态染色
细胞接种于激光共聚焦专用培养皿,实验结束后按照内质网形态染色试剂盒说明操作。染色后细胞在激光扫描共聚焦显微镜(FV1000, Olympus)下观察并采集图像。
10统计学处理
采用SPSS 13.0统计软件对实验数据进行分析,数据用均数±标准差(mean±SD) 表示,多组间比较应用单因素方差分析(One-way ANOVA),组间两两比较应用Student-Newman-Keuls检验,以P<0.05为差异有统计学意义。
结 果
1内质网应激诱导MVECs损伤
采用不同浓度内质网应激诱导剂TG作用培养MVECs,培养基LDH活性和细胞凋亡率测定检测TG对MVECs损伤的影响,结果发现TG剂量依赖性地诱导培养基LDH活性和细胞凋亡率增加,见图1。与正常对照组相比,0.1 μmol/L TG 诱导MVECs培养基LDH活性增加55.6% (P<0.05),但细胞凋亡率无显著变化。0.2 μmol/L TG 诱导MVECs凋亡率较正常对照组增加11.8% (P<0.05),随着TG浓度增加,LDH活性和细胞凋亡率剂量依赖性地增加。此外,1 μmol/L TG诱导MVECs损伤较为严重,培养基LDH活性和细胞凋亡率较0.5 μmol/L TG组分别增加29.5% (P<0.05)和11.1% (P<0.05)。故采用0.5 μmol/L TG 组诱导MVECs损伤进行下述实验。
2HPC减轻内质网应激诱导的MVECs损伤
采用0.5 μmol/L TG诱导MVECs显著损伤,低氧30 min复氧24 h进行HPC后观察MVECs损伤变化,结果发现内质网应激诱导MVECs显著损伤,TG组培养基LDH活性和细胞凋亡率分别较对照组增加70.3%(P<0.05)和12.37%(P<0.05),HPC可显著抑制TG诱导的微血管内皮细胞损伤,HPC+TG组LDH活性和细胞凋亡率分别较TG组降低21.6%(P<0.05)和8.58%(P<0.05),并且细胞凋亡率与对照组无显著差异(P>0.05)。单纯HPC对MVECs无显著损伤,见图2。

Figure 1. Effect of different doses of TG on MVECs injury. A:LDH activity in MVECs medium; B: apoptotic rate of MVECs. Mean±SD.n=4.*P<0.05vs0 μmol/L;#P<0.05vs0.5 μmol/L.
图1不同浓度TG对MVECs损伤的影响

Figure 2. Effect of HPC on TG-induced MVECs injury.A: LDH activity in MVECs medium; B: apoptotic rate of MVECs. Mean±SD.n=4.*P<0.05vscontrol;#P<0.05vsTG.
图2HPC对TG诱导的MVECs损伤的影响
3HPC减轻内质网应激诱导的MVECs通透性增加和细胞骨架破坏
FITC-albumin 荧光标记技术发现,内质网应激诱导MVECs通透性显著增加,TG组细胞通透性较对照组增加92.5% (P<0.05),HPC显著降低TG诱导的细胞通透性增加,HPC+TG组细胞通透性较TG组降低13.9% (P<0.05),但细胞通透性仍较对照组增加65.8%(P<0.05)。单纯HPC对微血管内皮细胞通透性无显著影响,见图3A。与MVECs通透性改变一致,内质网应激诱导MVECs细胞骨架显著破坏,TG组肌原纤维断裂,排列紊乱,无明显的应力纤维,细胞骨架内出现空泡。HPC显著减轻TG诱导的细胞骨架破坏,肌原纤维排列整齐,无显著断裂和空泡形成,与对照组无显著差异,见图3B,提示HPC通过减轻细胞骨架破坏进而发挥细胞保护作用。

Figure 3. Effects of HPC on permeability and cytoskeletal structure in MVECs treated with TG.A: permeability of MVECs; B: cytoskeletal structure in MVECs. Mean±SD.n=4.*P<0.05vscontrol;#P<0.05vsTG.
图3HPC对TG诱导的MVECs通透性和细胞骨架的影响
4TG诱导MVECs蛋白质表达谱变化
双向电泳-质谱技术发现TG作用后MVECs中7种蛋白质出现明显差异表达,与正常对照组比较,TG组6种蛋白质表达上调,1种蛋白质表达下调。7种蛋白质的序列号、蛋白名称和主要功能见表2。其中TG诱导MVECs细胞内质网应激分子GRP78和蛋白质二硫键异构酶相关蛋白3(protein disulfide isomerase-associated 3,PDIA3)显著升高,其差异图像和肽质量指纹图谱见图4。

Figure 4. Proteomic profiles of MVECs treated with or without TG.A: differential expression of PDIA3 and GRP78 in MVECs; B: peptide mass fingerprint of PDIA3;C: peptide mass fingerprint of GRP78.
图4TG诱导的MVECs蛋白质谱变化
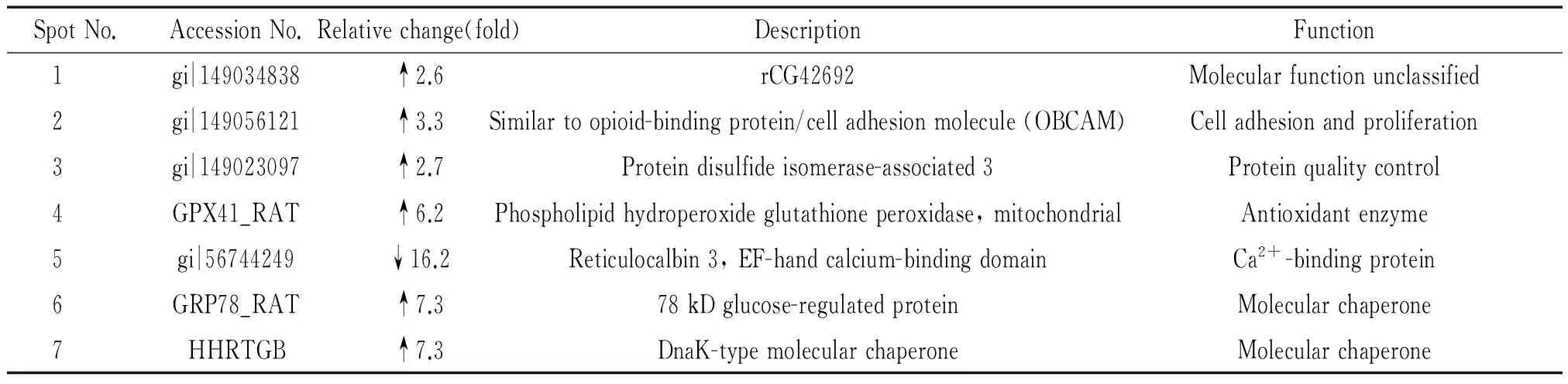
表2 质谱分析后7个蛋白点Mascot检索结果
↑: increased in TG groupvscontrol group; ↓: decreased in TG groupvscontrol group.
5HPC抑制MVECsCHOP介导的内质网应激相关凋亡途径
5.1内质网形态的改变 采用内质网特异性染料Dapoxyl标记MVECs内质网,激光共聚焦显微镜观察显示正常MVECs 内质网均匀分布在核周,无空泡出现;内质网应激诱导剂TG诱导MVECs 内质网形态显著受损,荧光颗粒分布不均、浓集,并有空泡形成;HPC显著抑制TG诱导的内质网损伤,荧光颗粒分布均匀,无空泡形成,与正常对照组无显著差异,见图5A。
5.2内质网应激分子CRT和GRP78表达的改变 采用RT-PCR和Western blotting技术检测内质网应激标志性分子CRT和GRP78的表达发现,TG诱导MVECs CRT和GRP78表达显著增加,TG组CRT mRNA和蛋白表达分别较对照组增加46.2% (P<0.05)和71.8% (P<0.05),GRP78 mRNA和蛋白表达分别较对照组增加46.7% (P<0.05)和44.4% (P<0.05)。HPC显著抑制CRT 蛋白表达,HPC+TG组CRT蛋白表达较TG组降低20.7% (P<0.05),但对CRT mRNA表达和GRP78 表达无显著抑制。单纯HPC可促进CRT表达增加,HPC组CRT mRNA和蛋白表达分别较对照组增加32.2% (P<0.05)和24.2%(P<0.05),但对GRP78 表达无显著影响,见图5B、C。
5.3CHOP相关凋亡途径变化 TG组CHOP mRNA和蛋白表达分别较对照组增加29.5%(P<0.05)和84.3%(P<0.05),HPC对TG诱导的CHOP mRNA表达无显著影响,但显著抑制TG诱导的CHOP表达增加,HPC+TG组CHOP蛋白水平较TG组降低35.6%(P<0.05)。单纯HPC对MVECs CHOP表达无显著影响,见图6。Bax是重要的促凋亡蛋白,而Bcl-2是重要的抑凋亡蛋白,Bax/Bcl-2比值反映HPC对TG诱导的MVECs损伤的影响。TG诱导的MVECs Bax/Bcl-2比值显著增加,TG组Bax/Bcl-2比值mRNA和蛋白水平分别较对照组增加181.5%(P<0.05)和107.5%(P<0.05),HPC显著抑制TG诱导的Bax/Bcl-2比值增加,HPC+TG组Bax/Bcl-2比值mRNA和蛋白水平分别较TG组降低76.1%(P<0.05)和40.4%(P<0.05)。单纯HPC对内皮细胞Bax/Bcl-2比值无显著影响,见图7。上述结果提示HPC可能通过抑制CHOP介导的内质网应激相关凋亡途径而发挥细胞保护作用。
讨 论
内皮功能障碍是高血压、动脉粥样硬化、冠心病等疾病的共同发病环节,决定疾病的发生、发展和疗效。心肌微血管内皮细胞在参与物质交换、防止血小板聚集和血栓形成等方面具有重要作用,而且通过分泌活性物质直接影响心肌细胞。微血管内皮细胞功能障碍和凋亡通过诱导动脉粥样硬化、血栓形成等多种作用参与了缺血性心脏病的发生。缺血预处理是迄今最强的内源性保护现象之一,并被HPC所模拟,自从1986年Murry等[9]发现这一现象以来,其对缺血及再灌注损伤的保护作用在多种模型和不同器官、组织上得到证实,但其内源性保护机制目前并未完全阐明,涉及减轻细胞内钙超载、氧化应激、诱导内源性保护蛋白合成等多种机制。内质网是细胞内稳态调节和蛋白质折叠、合成的细胞器,缺血、缺氧、钙超载、氧化应激等因素均可诱导ERS。大量研究证实ERS及相关凋亡参与了缺血性心脏病的发生、发展过程[3,10]。为证实HPC的内源性保护作用涉及直接减轻内质网应激诱导的细胞损伤,本工作在ERS诱导剂TG致大鼠心肌MVECs内质网应激模型上研究HPC的细胞保护作用,结果发现HPC显著抑制TG诱导的MVECs LDH漏出和细胞凋亡,减轻其通透性增加和细胞骨架破坏,提示HPC可以直接抑制内质网应激诱导的MVECs损伤。
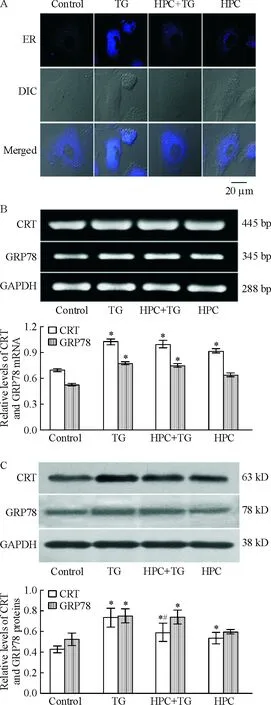
Figure 5. Effects of HPC on ER structure and expression of CRT and GRP78 in MVECs treated with TG.A: ER structure;B: mRNA levels of CRT and GRP78; C: protein levels of CRT and GRP78.DIC: differential interference contrast microscopy. Mean±SD.n=4.*P<0.05vscontrol;#P<0.05vsTG.
图5HPC对TG诱导的MVECsER形态以及CRT和GRP78表达的影响

Figure 6. Effect of HPC on CHOP expression in MVECs treated with TG.A: mRNA level of CHOP; B: protein level of CHOP. Mean±SD.n=4.*P<0.05vscontrol;#P<0.05vsTG.
图6HPC对TG诱导的MVECs内CHOP表达的影响
内质网应激通常表现为内质网分子伴侣CRT和GRP78表达上调。CRT是内质网内驻留的钙结合蛋白,在细胞钙稳态调节中具有重要作用[10],并与GRP78一起作为分子伴侣,调节蛋白合成、折叠[11],旨在稳定细胞内质网功能,保持细胞内环境稳态。PDIA3 是内质网应激蛋白成员,可被氧化应激诱导,参与蛋白质质量控制过程[12]。本实验采用双向电泳-质谱技术发现TG诱导MVECs内质网应激分子GRP78和PDIA3表达增加,Western blotting证实内质网应激分子TG诱导MVECs内CRT和GRP78表达显著增加,而内质网染色证实TG诱导MVECs内质网形态显著损伤和空泡化,上述结果提示TG诱导MVECs严重的内质网应激。

Figure 7. Effects of HPC on expression of Bax and Bcl-2 in MVECs treated with TG.A: mRNA level of Bax and Bcl-2; B: protein level of Bax and Bcl-2.Mean±SD.n=4.*P<0.05vscontrol;#P<0.05vsTG.
图7HPC对TG诱导的MVECs内Bax和Bcl-2表达的影响
研究发现CRT过度表达参与了大鼠冠状动脉结扎心肌梗死和培养心肌细胞缺氧复氧损伤,HPC诱导CRT适度表达参与了HPC的心肌保护作用,并抑制缺氧复氧诱导的心肌细胞CRT过表达和游离Ca2+浓度增加[11]。研究证实细胞内游离Ca2+浓度增加激活钙依赖性蛋白酶,继而降解收缩蛋白,引起细胞骨架破坏[13]。本实验发现内质网应激诱导MVECs内质网形态损伤和空泡化,细胞骨架严重破坏,无明显应力纤维,细胞骨架呈现弥散性破坏并出现空泡,同时细胞通透性显著升高。HPC显著抑制内质网应激诱导的上述损伤,内质网形态和细胞骨架与正常对照组无显著差异,提示HPC可能通过抑制CRT过表达,继而抑制细胞内Ca2+浓度增加诱导的骨架破坏而发挥细胞保护作用。
过度或严重内质网应激诱导内质网相关凋亡蛋白CHOP表达上调。CHOP又称为生长停滞及DNA损伤诱导蛋白153(growth arrest and DNA damage inducible protein 153, GADD153),它通过直接调节核中靶基因,增加细胞对内质网应激介导凋亡的敏感性。siRNA 干扰CHOP表达可抵抗内质网应激诱导的细胞凋亡[14],而CHOP过表达则促进细胞凋亡[15]。研究发现CHOP过表达导致抗凋亡蛋白Bcl-2表达降低,并且导致促凋亡蛋白Bax从胞浆内向线粒体内易位[16],Bcl-2过表达和bax基因敲除均可抑制CHOP诱导的细胞凋亡[17-18]。本实验发现内质网应激诱导剂TG诱导MVECs CHOP蛋白表达和Bax/Bcl-2比值显著增加,HPC显著抑制CHOP介导的细胞凋亡途径,提示HPC通过抑制内质网应激相关CHOP凋亡途径、减轻细胞凋亡继而发挥细胞保护作用。
本研究在原代培养心肌MVECs上发现内质网应激诱导MVECs显著损伤,促进细胞骨架破坏和通透性增加;HPC可能通过诱导适度内质网应激、抑制CHOP介导的内质网应激相关凋亡途径而减轻MVECs损伤及细胞骨架破坏,提示HPC的内源性保护机制可能涉及直接抑制CHOP介导的过度内质网应激反应,为内皮功能障碍相关疾病的防治提供了新的治疗靶点。
[1] Simionescu M. Implications of early structural-functional changes in the endothelium for vascular disease [J]. Arterioscler Thromb Vasc Biol, 2007, 27(2): 266-274.
[2] Liu XH, Wu XD, Cai LR, et al. Hypoxic preconditioning of cardiomyocytes and cardioprotection: phosphorylation of HIF-1α induced by p42/p44 mitogen-activated protein kinases is involved[J]. Pathophysiology, 2003, 9(4): 201-205.
[3] 张振英,刘秀华. 血管内皮细胞内质网应激[J].生理科学进展,2010, 41(1): 295-298.
[4] 刘秀华, 苏静怡. 缺血预处理的研究现状[J]. 生理科学进展,2001, 32(1): 83-87.
[5] Liu XH, Wu XD, Cai LR, et al. Calreticulin downregulation is associated with FGF-2-induced angiogenesis through calcineurin pathway in ischemic myocardium [J]. Shock,2008, 29(1): 140-148.
[6] Wu XD, Liu XH, Zhu XM, et al. Hypoxic preconditioning induces delayed cardioprotection through p38 MAPK-mediated calreticulin upregulation [J]. Shock, 2007, 27(5): 572-577.
[7] Gillrie MR, Krishnegowda G, Lee K, et al. Src-family kinase-dependent disruption of endothelial barrier function byPlasmodiumfalciparummerozoite proteins [J]. Blood, 2007, 110(9): 3426-3435.
[8] Zhang ZY, Liu XH, Hu WC, et al. The calcineurin-myocyte enhancer factor 2c pathway mediates cardiac hypertrophy induced by endoplasmic reticulum stress in neonatal rat cardiomyocytes[J]. Am J Physiol Heart Circ Physiol,2010, 298(5): H1499-H1509 .
[9] Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium [J]. Circulation, 1986, 74(5): 1124-1136.
[10] Park K, Poburko D, Wollheim C, et al. Amiloride derivatives induce apoptosis by depleting ER Ca2+stores in vascular endothelial cells [J]. Br J Pharmacol, 2009, 156(8):1296-1304.
[11] Liu XH, Xu FF, Fu Y, et al. Calreticulin induces delayed cardioprotection through mitogen-activated protein kinases [J]. Proteomics,2006, 6(13): 3792-3800.
[12] Huang TS, Olsvik PA, Krvel A, et al. Stress-induced expression of protein disulfide isomerase associated 3 (PDIA3) in Atlantic salmon (SalmosalarL.) [J]. Comp Biochem Physiol B Biochem Mol Biol,2009, 154(4): 435-442.
[13] Papp Z, van der Velden J, Stienen GJ. Calpain-I induced alterations in the cytoskeletal structure and impaired mechanical properties of single myocytes of rat heart [J].Cardiovasc Res,2000, 45(4): 981-993.
[14] Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress [J]. Cell Death Differ, 2004, 11(4): 381-389.
[15] Kim KM, Pae HO, Zheng M, et al. Carbon monoxide induces heme oxygenase-1 via activation of protein kinase R-like endoplasmic reticulum kinase and inhibits endothelial cell apoptosis triggered by endoplasmic reticulum stress [J]. Circ Res, 2007, 101(9): 919-927.
[16] Friedman AD. GADD153/CHOP, a DNA damage-inducible protein, reduced CAAT/enhancer binding protein activities and increased apoptosis in 32D c13 myeloid cells [J]. Cancer Res,1996, 56(14): 3250-3256.
[17] McCullough KD, Martindale JL, Klotz LO, et al. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state [J]. Mol Cell Biol,2001, 21(4): 1249-1259.
[18] Gotoh T, Terada K, Oyadomari S, et al. hsp70-DnaJ chaperone pair prevents nitric oxide-and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria [J]. Cell Death Differ,2004, 11(4): 390-402.
Hypoxicpreconditioningprotectsratmyocardialmicrovascularendothelialcellsagainstendoplasmicreticulumstress-inducedinjury
WU Xu-dong1, ZHANG Zhen-ying2, WANG Xiao-reng2, LI Yu-zhen2, LIU Xiu-hua2
(1Out-patientDepartment,2DepartmentofPathophysiology,ChinesePeople’sLiberationArmyGeneralHospital,Beijing100853,China.E-mail:wuxudong60@yahoo.com.cn)
AIM: To investigate the effect of hypoxic preconditioning (HPC) on endoplasmic reticulum stress (ERS)-induced injury in cultured microvascular endothelial cells (MVECs) from rat hearts.METHODSMVECs injury was induced by an ERS inductor thapsigargin (TG). Lactate dehydrogenase (LDH) leakage and apoptotic rate were detected to evaluate the injury of MVECs. Cytoskeleton and endoplasmic reticulum (ER) in MVECs were observed by phalloidin-FITC fluorescence staining and ER staining, respectively. Two-dimensional electrophoresis and mass spectrometry (MS) were used to identify proteomic profile in MVECs treated with TG. Western blotting was used to detect the expression of ERS markers, calreticulin (CRT) and glucose-regulated protein 78 (GRP78).RESULTSTG induced the increase in LDH activity in medium and the apoptosis of MVECs in a dose-dependent manner. TG treatment up-regulated the expression of CRT and GRP78, while HPC attenuated the ERS-induced injury and the up-regulation of ERS markers in MVECs.CONCLUSIONHPC protects MVECs from ERS-induced injury.
Hypoxic preconditioning; Microvascular endothelial cells; Endoplasmic reticulum stress
R363.1
A
1000- 4718(2013)09- 1537- 09
2012- 09- 17
2013- 08- 16
国家自然科学基金资助项目(No. 81070186)
△通讯作者 Tel: 010-66937334; E-mail: wuxudong60@yahoo.com.cn
▲并列第1作者
10.3969/j.issn.1000- 4718.2013.09.001
