复方刺山柑胶囊的质量标准研究
2013-09-17杨伟俊罗玉琴薛文采
袁 涛 杨伟俊 罗玉琴 薛文采 程 波
1.新疆维吾尔自治区克拉玛依市中心医院,新疆克拉玛依 834000;2.新疆维吾尔自治区药物研究所新疆维吾尔药重点实验室,新疆乌鲁木齐 830004;3.中国科学院新疆理化技术研究所 中国科学院干旱区植物资源化学重点实验室,新疆乌鲁木齐 830011
复方刺山柑胶囊是通过临床经验结合现代药物配伍筛选技术自主研发的复方药物,处方由刺山柑、川西獐牙菜、波棱瓜子、大黄、苦荬菜、木香、兔耳草、唐古特乌头、角茴香、甘草、金钱草、诃子、黄芪等多味药材组成,具有清热解毒、疏肝利胆、利湿退黄之功能,用于急性黄疸型肝炎、急性胆囊炎属肝胆温热证者。课题组前期根据处方各药味所含化学成分的理化性质结合本方功效,优化了复方刺山柑胶囊的提取工艺[1],并制备成胶囊剂。本文此其基础上进行制剂的质量研究,为临床用药安全和有效提供质量控制标准。
1 材料与仪器
1.1 试验材料
复方刺山柑胶囊(批号:100402、100403、100404、100405、100415、100416、100417、100723、100724);大黄素(中国药品生物制品检定所提供,批号:0756-200110);大黄酚对照品(中国药品生物制品检定所提供,含量测定用,批号:0796-200208);齐墩果酸对照品 (中国药品生物制品检定所提供,含量测定用,批号:110709-201206);黄芪甲苷对照品(中国药品生物制品检定所提供,含量测定用,批号:110781-200613);没食子酸对照品(中国药品生物制品检定所提供,含量测定用,批号:110831-200803);木香对照药材(中国药品生物制品检定所提供,批号:120921-201008);诃子对照药材 (中国药品生物制品检定所提供,批号:121015-201004);川西獐牙菜对照药材(中国药品生物制品检定所提供,批号:121354-200401);大黄对照药材(中国药品生物制品检定所提供,批号:120984-201202)。薄层层析用硅胶G(化学纯,青岛海洋化工有限公司);甲醇为色谱纯(美国Fisher公司),其他试剂均为分析纯。
1.2 仪器设备
Shimadzu-LC 2010C全自动液相色谱仪,Shimadzu CLASS-VP V6.14SP1数据工作站 (日本岛津制造所);LibrorAEG-200电子天平(日本岛津制造所);WHF-203B暗箱式三用紫外分析仪 (上海精科实业有限公司);Milliporesimplicity-185超纯水器 (美国密理博公司);KQ-100DE型数控超声波发生器(昆山市超声仪器有限公司)。
2 薄层色谱鉴别
2.1 川西獐牙菜的鉴别
取本品内容物5 g,加乙醇30 mL,超声处理20 min,滤过,滤液加水15 mL和盐酸2 mL,加热回流1 h,放冷,加石油醚(60~90℃)40 mL振摇提取,提取液蒸干,残渣加乙醇2 mL使溶解,作为供试品溶液。另取川西獐牙菜对照药材2 g,加乙醇20 mL,加热回流1 h,滤过,同法制成对照药材溶液。再取齐墩果酸对照品,加乙醇制成每毫升含1 mg的溶液,作为对照品溶液。参照薄层色谱法(《中国药典》2010年版一部附录ⅥB)试验,点样量均为10 μL,采用硅胶G薄层板,展开剂为环己烷-丙酮-乙酸乙酯(5∶2∶1),展开,取出,晾干,显色剂为10%的硫酸乙醇溶液,105℃加热显色。在与对照药材和对照品色谱相应的位置上,供试品色谱显相同颜色的斑点。
2.2 大黄的鉴别
取本品内容物2 g,加20 mL甲醇,超声处理15 min,滤过,滤液挥干,用20 mL水,溶解后加盐酸2 mL,加热回流20 min,立即冷却,用乙醚振摇提(2次 ×20 mL),合并乙醚液,蒸干,用1 mL三氯甲烷溶解后即为供试品溶液。取大黄对照药材0.5 g,按供试品溶液制备方法处理成对照药材溶液。再取大黄酸对照品,加甲醇制成每毫升含1 mg的溶液,作为对照品溶液。参照薄层色谱法(《中国药典》2010年版一部附录ⅥB)试验,点样量均为5 μL,采用硅胶G薄层板,展开剂为石油醚(30~60℃)-甲酸乙酯-甲酸(15∶5∶1)的上层溶液,展开,取出,晾干,置365 nm紫外光灯下检视。供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同的橙黄色荧光斑点;置氨蒸气中熏后,斑点变为红色。
2.3 黄芪的鉴别
取本品内容物6 g,加甲醇80 mL,加热回流1 h,放冷,滤过,滤液蒸干,残渣加水50 mL使溶解,滤过,滤液用水饱和的正丁醇振摇提取 (3次×40 mL),合并正丁醇液,用氨试液洗涤2次,每次50 mL,取正丁醇液,再用正丁醇饱和的水洗涤(2次×50 mL),正丁醇液蒸干,残渣加水5 mL使溶解,放冷,通过D101型大孔吸附树脂(内径1.5 cm,长12 cm),以水80 mL洗脱,弃去水液,再用40%乙醇50 mL洗脱,弃去洗脱液,继用70%乙醇100 mL洗脱,收集洗脱液,蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液。另取黄芪甲苷对参照品,加甲醇制成每毫升含1 mg的溶液,作为对照品溶液。照薄层色谱法(《中国药典》2010年版一部附录ⅥB)试验,点样量均为10 μL,采用硅胶G薄层板,展开剂为三氯甲烷-乙酸乙酯-甲醇-水(15∶40∶22∶10)10℃以下放置的下层溶液,显色剂为10%硫酸乙醇溶液,105℃加热显色。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。
2.4 木香的鉴别
取本品内容物3 g,加乙醚15 mL,振摇5 min,放置2 h,滤过,滤液作为供试品溶液。另取木香对照药材1 g,加乙醇10 mL,同法制成对照药材溶液。参照薄层色谱法(《中国药典》2010年版一部附录ⅥB)试验,点样量均为10 μL,采用硅胶G 薄层板,展开剂为环己烷-丙酮(10∶3),展开,取出,晾干,显色剂为5%香草醛硫酸溶液,105℃加热显。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点。
2.5 诃子的鉴别
取本品内容物2 g,加乙醇10 mL,超声处理10 min,放置30 min,滤过,滤液作为供试品溶液。另取诃子对照药材1 g,加乙醇5 mL,同供试品溶液制备方法制成对照药材溶液。再取没食子酸对照品,加乙醇制成每毫升含1 mg的溶液,作为对照品溶液。参照薄层色谱法(《中国药典》2010年版一部附录Ⅵ B)试验,点样量均为5 μL,采用硅胶 GF254 薄层板,展开剂为三氯甲烷-乙酸乙酯-甲酸(5∶4∶1),展开,取出,晾干,置254 nm紫外光灯下检视。供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的荧光斑点。
3 大黄素和大黄酚含量测定[2]
3.1 色谱条件
Kromasil ODS-l色谱柱 (250 mm× 4.6 mm,5 μm);甲醇-0.1%磷酸溶液(85∶15)为流动相;柱温:30℃;以 254 nm为检测波长。
3.2 对照品溶液的制备
精密称取大黄素、大黄酚对照品各5 mg,分别置50 mL量瓶中,用甲醇溶解并稀释至刻度,摇匀;分别精密吸取大黄素溶液1 mL、大黄酚溶液2 mL,分别置50 mL量瓶中,加甲醇至刻度,摇匀,即得(大黄素每1 mL中含2 μg,大黄酚每毫升中含 4 μg)。
3.3 供试品溶液的制备
取本品约1.0 g,精密称定,置100 mL锥形瓶中,加8%盐酸20 mL,超声处理5 min,再加三氯甲烷20 mL,加热回流1 h,冷却,移置分液漏斗中,用少量三氯甲烷洗涤容器,并入分液漏斗中,分取三氯甲烷层,酸液用三氯甲烷提取(2次×10 mL),合并三氯甲烷液,置水浴上挥干溶剂,残渣加适量甲醇微热使溶解,移入50 mL量瓶中,用少量甲醇分次洗涤容器,洗液移入同一量瓶中,加甲醇至刻度,摇匀,用微孔滤膜(0.45 μm)滤过,取续滤液,即得。
3.4 阴性对照溶液的制备
按本品处方比例称取缺大黄的其余药味,按生产工艺制成阴性对照,取阴性对照细粉约1 g,精密称定,置100 mL具塞锥形瓶中,依供试品溶液制备方法项下自“加8%盐酸20 mL……”起,制备阴性对照溶液。
在上述条件下,各蒽醌类成分与供试品溶液中其他色谱峰达到了基线分离,阴性对照试验结果表明无干扰,色谱图见图1。
3.5 线性关系考察
3.5.1 大黄素线性范围考察 精密称取大黄素对照品适量,加甲醇制成每毫升含0.096 mg的溶液,摇匀,精密量取1 mL置25 mL量瓶中,加甲醇稀释至刻度,摇匀,作为贮备液。分别精密量取贮备液1、2、3、4 mL置5 mL量瓶中,加甲醇稀释至刻度,摇匀,分别制成每毫升含0.768、1.536、2.304、3.072 μg的溶液。分别精密吸取上述梯度浓度的对照品溶液与贮备液10 μL,注入液相色谱仪,记录峰面积。以进样浓度C(μg/mL)为横坐标,峰面积A为纵坐标进行线性回归,得标准曲线方程为:A=33319 C+1536.1,r=0.9996。表明在0.768~3.840 μg/mL范围内,大黄素峰面积与进样浓度具有良好的线性关系。
3.5.2 大黄酚线性范围考察 精密称取大黄酚对照品适量,加甲醇制成每毫升含0.102 mg的溶液,作为贮备液。分别精密量取贮备液 0.5、1、2、3、4 mL 置 25 mL量瓶中, 加甲醇分别制成每毫升含 2.04、4.08、8.16、12.24、16.32 μg 的溶液。分别精密吸取上述溶液各10 μL,注入液相色谱仪,记录峰面积。以进样浓度C(μg/mL)为横坐标,峰面积A为纵坐标进行线性回归,得标准曲线方程为:A=39750 C+22239,r= 0.9993。表明在 2.04~16.32 μg/mL 范围内,大黄酚峰面积与进样浓度具有良好的线性关系。
3.6 稳定性实验
同一批样品(批号:100402)按照上述供试品制备方法制成供试品溶液,在上述色谱条件下,每隔2 h进样1次,每次10 μL,计算大黄素和大黄酚总量的RSD为0.49%。
3.7 精密度
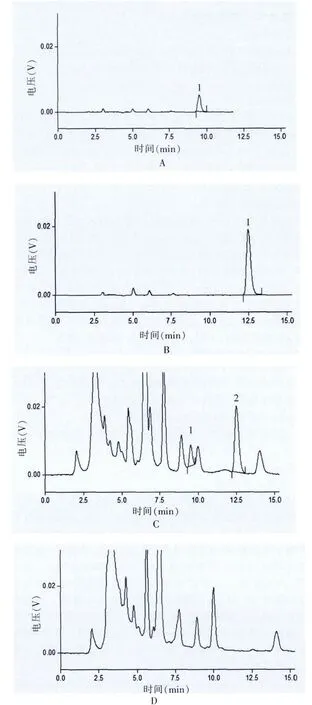
图1 复方刺山柑胶囊中大黄HPLC图谱
3.7.1 重复性 取同一份供试品溶液,在上述色谱条件下重复进样5次,峰面积的RSD:大黄素为1.56%,大黄酚为1.24%。
3.7.2 日间精密度 取同一批样品(批号:100402)分别在不同天(连续5 d),按照上述供试品制备方法制成供试品溶液,在上述色谱条件下测定,计算大黄素和大黄酚总量的RSD为0.70%。
3.8 回收率测定
3.8.1 大黄素 精密称取供试品细粉0.5 g,共10份,其中一组为空白组,其余分三组,分别加入0.096 mg/mL的大黄素对照品溶液0.31、0.62、1.05 mL,照供试品溶液制备方法处理后,在上述色谱条件下进样10 μL,计算回收量和回收率,结果表明,9次测定的平均回收率为100.00%(n=9),RSD 为 1.18%(n=9)。见表 1。
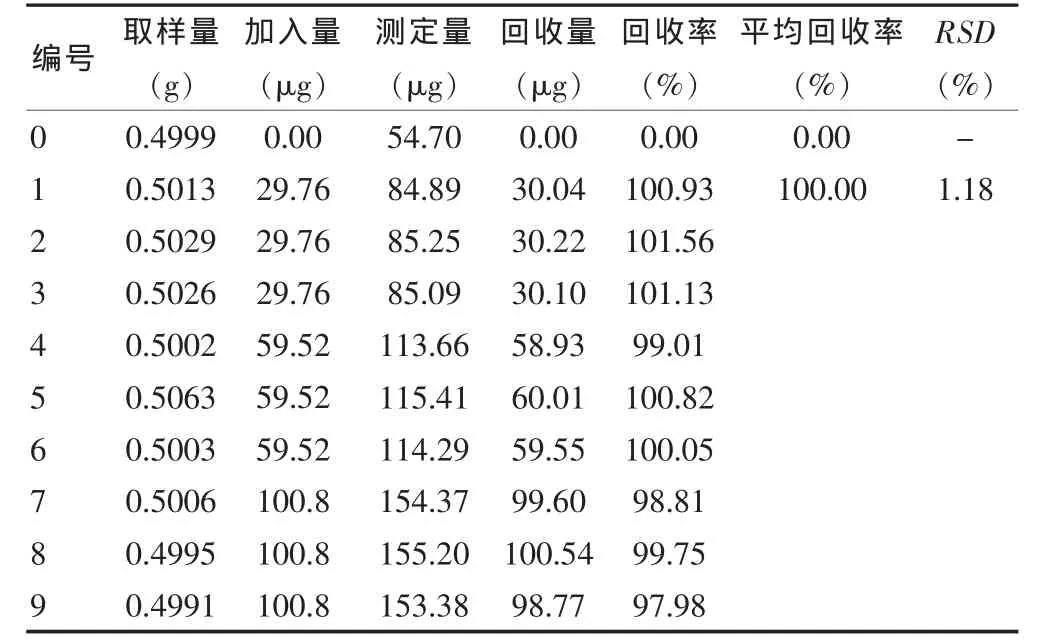
表1 大黄素回收率测定结果
3.8.2 大黄酚 精密称取供试品细粉0.5 g,共10份,其中一组为空白组,其余分三组,分别加入0.102 mg/mL的大黄酚对照品溶液1、2、3.5 mL,照供试品溶液制备方法处理后,在上述色谱条件下进样10 μL,计算回收量和回收率,9次测定的平均回收率为100.62%(n=9),RSD 为 0.38%(n=9)。见表2。
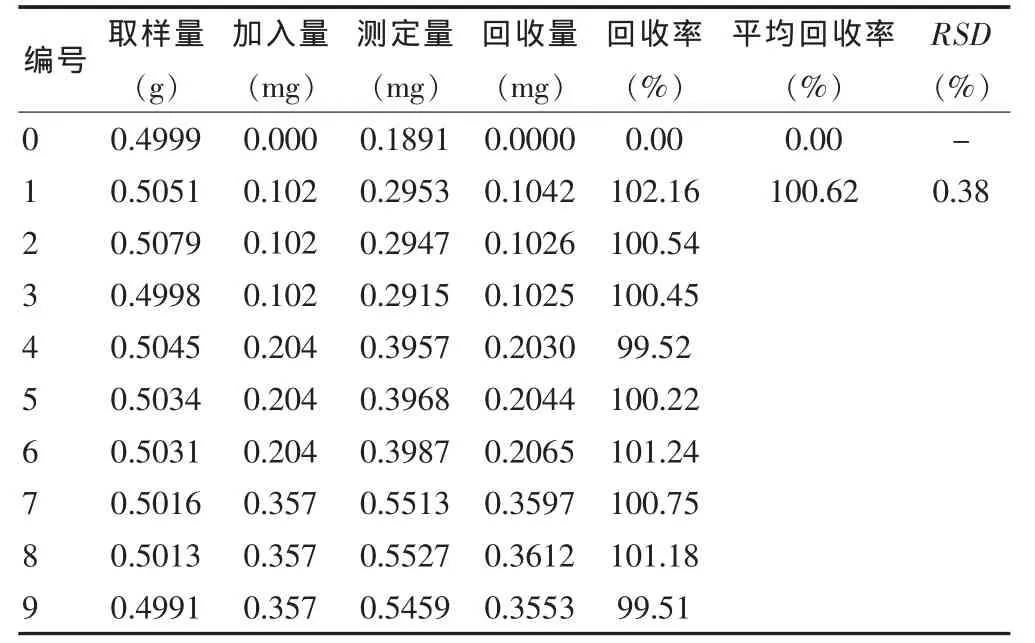
表2 大黄酚回收率测定结果
3.9 10批样品测定
对制备的10批样品(其中3批为中试规模)进行含量测定,每批样品重复测定2次,计算平均值,结果见表3。
考虑到药材的产地来源,以及制剂生产、储藏等因素,暂定本品每克含大黄以大黄素和大黄酚总量计,不得低于0.35 mg。按每粒装0.36 g计算,限度0.13 mg/粒。
4 讨论
4.1 质量控制指标的选择依据
选择合理的质量控制指标是中药复方药物研究的关键点,也是难点,而将工艺筛选研究中的考察指标纳入质量控制体系不仅能够追踪药效成分或化学成分的转移过程,而且有利于验证工艺路线的稳定性和可行性。本品工艺研究中,有学者采用80%乙醇提取了川西獐芽菜、大黄、木香等药味,其总蒽醌提取率达13.09~17.62 mg/g,正丁醇萃取物得率为13.31~16.32 mg/g,三氯甲烷萃取物得率达7.22~9.64 mg/g[1]。本文通过对大黄中两种蒽醌类成分的测定,计算其转移率达65%以上,与工艺研究结果吻合。川西獐芽菜[3-5]、大黄[6]、木香[7]中脂溶性成分具有保肝利胆作用,通过定性研究,对脂溶性有效成分进行了有效控制。针对诃子[8]、黄芪[9-10]中含大量多糖和鞣质类成分及与本方功能主治紧密结合的特点,提取工艺采用复方共煎,本文对二味进行TLC鉴别研究。本研究兼顾了提取工艺的考察指标,最大限度地保证了临床的安全有效和质量可控。

表3 10批样品中大黄素和大黄酚总量测定结果(mg/g)
4.2 最小检测限、定量限测定
根据复方中大黄素和大黄酚含量较低的特点,研究中分别配制浓度适当的大黄素和大黄酚对照品溶液,在含量测定色谱条件下进样10 μL,结果表明大黄素的最小检测浓度约 0.261 μg/mL(信噪比 S/N=3),定量浓度约 0.384 μg/mL(信噪比S/N=10);大黄酚的最小检测浓度约0.163 μg/mL(信噪比 S/N=3),定量浓度约 0.408 μg/mL(信噪比 S/N=10)。检测结果表明本品测定浓度远高于两种含量测定指标的定量限,为定量测定提供了依据。
中药复方的成分复杂,采用几个指标成分或药效成分进行定性定量研究难以全面控制产品质量,课题组对处方中刺山柑的化学成分和质量标准进行了研究[11-12],同时对其他药味均进行了质量控制研究,因专属性不强或无对照物质,无法作为质量控制指标。今后还需要深入研究其药效物质基础,建立能够全面控制本品质量的标准。
[1]焦胜春,杨飞,杨伟俊,等.复方刺山柑胶囊提取工艺的优化[J].中成药,2011,33(12):2169-2171.
[2]国家药典委员会.中国药典[S].一部.北京:中国医药科技出版社,2010:22-23.
[3]尚军,张国燕,杨淳彬,等.川西獐牙菜的化学成分研究[J].青海师范大学学报自然科学版,2008,(4):66-67.
[4]罗桂花,赵建平,陈海娟,等.川西獐牙菜醇提物对小鼠CCl4肝损伤的保护作用[J].四川中医,2008,26(11):29-30.
[5]吕坪,杜玉枝,李岑,等.川西獐牙菜醇提水沉部位抗黄疸性肝损伤的活性研究[J].时珍国医国药,2011,22(5):1098-1099.
[6]王耀先,郝慧南,陈秀玮,等.大黄素通过内源性线粒体途径和外源性死亡受体途径诱导人宫颈癌Hela细胞凋亡的研究[J].中国中医药科技,2013,(1):37-39.
[7]许丽佳,章津铭,瞿燕,等.川木香醇提物利胆镇痛作用的实验研究[J].江苏中医药,2010,42(9):76-77.
[8]Harpreet W,Rajbir S,Subodh K,et al.Effect of fractionation on antiradical efficacy of ethyl acetate extract of Terminalia chebula Retz[J].African Journal of Pharmacy and Pharmacology,2010,4(5):276-285.
[9]吴强,杨雁,薛绍礼,等.黄芪总苷对肝星状细胞增殖和合成胶原的抑制作用[J].中国药理学通报,2003,19(8):892.
[10]马莹,李润琴,贾建伟,等.注射用黄芪多糖联合肝动脉栓塞化疗治疗原发性肝癌疗效观察[J].中草药,2008,39(12):1856-1858.
[11]赵军,杨伟俊,任远,等.刺山柑化学成分研究[J].天然产物研究与开发,2012,24(1):52-54,24.
[12]杨伟俊,阿不都沙拉木,陈燕,等.刺山柑果质量标准研究[J].时珍国医国药,2011,22(1):133-134.
