中国人群低钾型周期性麻痹家系CACNA1S和SCN4A基因突变状态分析
2013-04-01武昆王晓英姚合斌
武昆,王晓英,姚合斌
低钾型周期性麻痹(hypokalemic periodic paralysis,HPP)包括家族性或散发性和甲状腺功能亢进性周期性麻痹,是常染色体显性遗传性肌肉病,以伴随血清钾降低的发作性肌无力为特征,肌无力经常累及四肢,严重者可死于呼吸肌麻痹或血清钾降低所致的心律失常[1-3]。近年来通过遗传学、分子生物学和电生理学的联合研究发现,HPP是由离子通道基因突变所致的离子通道病,涉及的离子通道基因包括编码骨骼肌电压门控钙通道α1亚单位基因(CACNA1S)和编码骨骼肌电压门控钠通道α亚单位基因(SCN4A)。1994年,Ptácek等[4]和Jurkat-Rott等[5]几乎同时报道了本病是由于二氢吡啶受体敏感的钙离子通道(L型钙通道)α1亚基(CACNA1S)的DNA序列点突变所致。此后1999年,Bulman等[6]在编码电压门控钠离子通道α亚单位的SCN4A基因上发现存在点突变(Arg669→His)。到目前为止,国内外研究者们在CACNA1S和SCN4A中已发现了多个突变位点。
本文针对2个家族性HPP家系、1个甲亢性HPP家系及4例散发性HPP患者,采用PCR和DNA直接测序技术对CACNA1S、SCN4A基因突变位点进行检测,并对相关文献进行回顾,以期在基因水平上为该病的发病机制和流行病学研究提供更多证据,现将结果报告如下。
1 资料与方法
1.1 研究对象 2个家族性HPP家系6例患者及其亲属,1个甲亢性HPP家系2例患者,4例散发性HPP患者。家族性HPP家系A(图1A),3代2例患者。家族性HPP家系B(图1B),3代4例患者。甲亢性HPP家系C(图1C),3代1例患者。所有患者均表现为突然发作的低血钾和周期性发作的骨骼肌迟缓性瘫痪。先证者均接受血生化、血气分析、甲状腺功能、甲状腺及肾上腺B超、尿常规等辅助检查。

图1 HPP家系Fig. 1 Pedigrees of HPP A. Pedigree A of a Chinese familial HPP; B. Pedigree B of a Chinese familial HPP; C. Pedigree C of a Chinese hyperthyroid HPP; □represents the healthy male; ○represents the healthy female; represents the male patient; ●represents the female patient; ↗represents the proband
1.2 基因检测方法 所有患者及其家属签署知情同意书后,取外周血3ml,应用试剂盒(WizardTMGenomic DNA Purification Kit,美国Promega)提取基因组DNA。自行设计CACNA1S基因外显子11、20、21、22、26、30及SCN4A基因外显子5、12、18、19引物(表1),利用PCR技术对外显子进行扩增,引物由奥科生物工程技术公司合成。PCR反应体系50μl,含DNA 50ng,引物0.2μmol/L,dNTP 100μmol/L,Taq DNA聚合酶2U(康为公司)。反应条件:94℃变性5min;94℃变性30s,55℃退火30s,72℃延伸30s,30个循环;延伸5min。应用2%琼脂糖电泳检测PCR产物,用Promega凝胶回收试剂盒(申能博采生物技术公司)回收PCR产物。以回收产物为模板再行PCR扩增,之后对PCR产物再次回收。用DNA测序仪(Prism 377,美国ABI)对回收后的PCR产物行正反向测序,明确有无突变。
1.3 文献检索及分析 检索PubMed数据库,搜集1999年1月-2012年12月公开发表的关于HPP家系CACNA1S、SCN4A基因突变的相关文献,关键词为“低钾型周期性麻痹”、“CACNA1S”、“SCN4A”、“突变”等,检索语种为英文。文献纳入标准:研究对象相同(不包括继发性HPP人群),研究方法相似,病例诊断标准相同,提供充足的原始资料。排除标准:重复发表的文献,未提供充足原始数据的文献,Meta分析类文献,综述类文献。

表1 CACNA1S和SCN4A基因待检测位点外显子PCR引物Tab. 1 PCR primers applied for detection of target exons in gene CACNA1S and SCN4A
2 结 果
2.1 诊断及临床资料 诊断标准参照文献[7]进行:根据常染色体显性遗传或散发,突发四肢弛缓性瘫痪,近端为主,无脑神经支配肌肉损害,无意识障碍和感觉障碍,数小时至1d内达高峰,辅助检查示血钾降低,心电图呈低钾性改变,经补钾治疗肌无力迅速缓解。
2个家族性HPP家系患者具有典型症状,结合家族史可以获诊断。另外1个甲亢性HPP家系的1例患者具有甲亢和HPP的临床表现,并经甲状腺功能测定证实甲状腺功能亢进,同时排除其他继发性因素,获得诊断。4例散发患者根据典型HPP症状,排除家族史及其他继发性原因,获得诊断。
病史和临床特征见表2。家系A:先证者(Ⅲ8),男,23岁。22岁开始出现迟缓性四肢无力,多由双下肢开始,数小时逐渐蔓延至双上肢。一般在晨起时发作,口服氯化钾缓释片(商品名:补达秀)治疗持续12~24h后肢体肌力可逐渐恢复正常,无肌强直表现。平时一直口服氯化钾缓释片和氯化钾口服液预防发作。发作诱因有劳累、受凉、寒冷、晚餐进食过饱、情绪紧张、酗酒、喝碳酸类饮料、运动后大量出汗等,寒冷可使症状加重。在起病初发作频率10-15次/年,23岁开始病情逐渐加重。发作期检查:血钾1.4~2.5mmol/L(平均1.9mmol/L);心电图示T波低平,U波增高;但多次查血气分析、尿常规、肾功能、甲状腺功能、甲状腺及肾上腺B超、肌电图均正常。

表2 HPP家系患者临床特征Tab. 2 Clinical features of patients with hypokalemic periodic paralysis
家系B:先证者(Ⅱ3),女,46岁,首发年龄40岁。一般在睡眠中发病,持续24h后肢体肌力恢复正常。诱因主要有劳累、受凉、出汗、饱食、月经前期等。发作频率:15~20次/年。发作期检查:血钾偏低(1.8mmol/L),尿常规、肌电图正常。补钾治疗有效,乙酰唑胺治疗无效。
家系C:先证者(Ⅱ7),男,48岁,首发年龄43岁。甲状腺功能亢进病史10年,一般在饮酒、劳累后发病,持续24h后肢体肌力恢复正常。诱因主要有劳累、受凉、出汗、饮酒等。发作频率∶10~15次/年。发作期检查:血钾偏低(2.5mmol/L),尿常规、肌电图正常。口服钾剂有效。
2.2 DNA测序结果 见图2、3、4、5。家系A共3代41人(检测33人,Ⅱ3、Ⅱ5、Ⅲ3、Ⅲ5失访),其中2例发病者为先证者及其爷爷,均为男性。先证者,男,23岁,首发年龄22岁;先证者爷爷,76岁,首发年龄56岁。本研究中,患者及其家族成员的PCR产物经测序未在目前已报道的突变位点中发现突变。家系B共3代26人(检测20人,Ⅱ2、Ⅱ6、Ⅱ10失访),其中4例发病。发病者中男性3例,女性1例。先证者为女性,46岁,首发年龄40岁。3例男性均是先证者的兄弟。本研究中,家系B中的患者及家族成员,其PCR产物经测序也未发现突变。家系C共3代16人(检测12人,Ⅱ2、Ⅱ4失访),其中1例发病。先证者,男,48岁,首发年龄43岁。本研究中,患者及其家族成员的PCR产物经测序未发现目前已知突变。4例散发性周期性麻痹患者的测序结果也无阳性发现。

图2 正常对照、已报道HPP突变位点及HPP家系先证者CACNA1S基因外显子11第528位氨基酸附近的序列图Fig. 2 Sequence map of amino acids at position 528 and nearby on exon 11 in gene CACNA1S of normal control, the reported mutation site of HPP and the proband of HPP pedigree

图3 正常对照及HPP先证者CACNA1S基因外显子11第1567至1596位碱基测序图Fig. 3 Sequence map of basic radicals ranging from 1567bp to 1596bp on exon 11 in gene CACNA1S of normal control and the proband of HPP pedigrees

图4 正常对照、已报道HPP突变位点及HPP家系先证者SCN4A基因外显子12第672位氨基酸附近的序列图Fig. 4 Sequence map of amino acids at position 672 and nearby on exon 12 in gene SCN4A of normal control, the reported mutation site of HPP and the proband of HPP pedigree
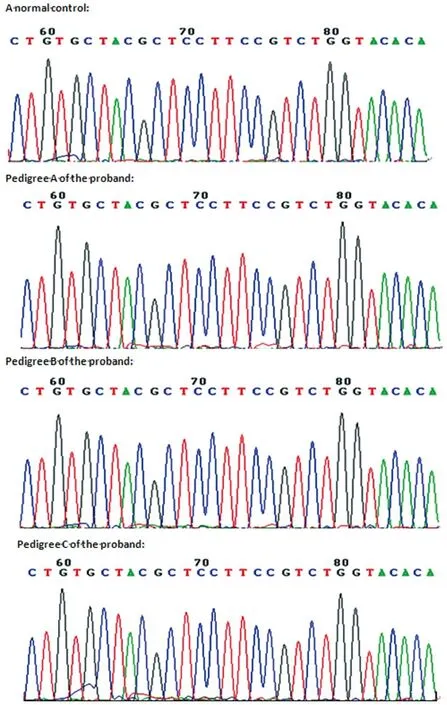
图5 正常对照及HPP家系先证者SCN4A基因12外显子1999至2028位碱基测序图Fig. 5 Sequence map of basic radicals ranging from 1999bp to 2028bp on exon 12 in gene SCN4A of normal control and the proband of HPP pedigrees
2.3 文献分析结果 检索到符合标准的文献92篇,按排除标准进行排除和筛选,最后纳入文献9篇,其中西方白种人群个案报道4篇、HPP家系基因突变文献5篇。在个案报道中3篇报道了CACNA1S突变[8-10],1篇报道了SCN4A突变[6];在HPP家系基因突变文献中,多是西方白种人群大样本研究,相关突变的基因也是以CACNA1S为主。除了检索相关西方白种人群的文献,对中国人群及东亚人群进行的相关检索发现,个案报道7篇[11-17],多为CACNA1S突变;家系基因突变的文献7篇[18-24],但样本量较小,多为单个家系研究。到目前为止,国内外研究者均在CACNA1S和SCN4A发现了多个突变位点(表3)。进一步分析发现,西方白种人群的基因突变以家族性为主,散发性突变较少;HPP突变家系的样本量大于中国人群及东亚人群;CACNA1S及SCN4A基因的突变阳性率为33.3%~87.9%。
3 讨 论
HPP是显性遗传性疾病,为周期性麻痹(periodic paralysis,PP)中最常见的类型,因各种因素影响使本病外显不全。本病的基因突变可能来自于父母或患者本人基因发生的突变。本研究患者父母及亲属无相应突变,因此,笔者推测家系A中第2代没有发病者,隔代出现了发病者的情况可能与患者本人基因突变有关。
本病以发作性肌无力伴发作期血钾降低、补钾后症状迅速缓解为特征。HPP发病以青壮年为多,多在睡眠中发病,临床主要表现为对称性双下肢无力,逐渐向上发展,以近端较重。发作时神志清楚,呼吸、吞咽、咀嚼、发音、眼球运动常不受影响,严重病例可累及呼吸肌甚至死亡[25]。不同患者的初发年龄、持续时间、严重程度及发作周期各异[26-27]。目前低钾型周期性麻痹主要通过临床病史、发病时血清钾浓度以及补钾治疗有效等情况进行诊断,而基因分析可协助临床诊断的确定。

表3 HPP患者CACNA1S和SCN4A基因已确认的突变位点Tab. 3 Verified mutation sites in patients with hypokalemic periodic paralysis
在目前西方人群大样本研究中,以Sternberg等[28]及Matthews等[29]所做的研究为代表。据Sternberg等[28]报道,在58个独立的家族性HPP家系中,突变的阳性率为77.6%。其中40个家系与CACNA1S基因突变有关,比例为69.0%,包括R528H突变和R1239H突变,分别占45.0%和24.0%;5个家系与SCN4A基因突变有关,比例为8.6%;其余13个家系无突变。2011年, Matthews等[29]报道在83个独立的HPP患者中突变阳性率为87.9%,其中65例(78.3%)为CACNA1S突变,8例(9.6%)为SCN4A突变。在东方人群中,目前报道的突变位点与西方基本相同,但CACNA1S及SCN4A基因突变阳性率低于西方人群,即使是与西方人群不同的突变位点,其突变也多是个案报道[17,19]。
在目前的突变位点中,主要是以R528H、R1239H为常见,其他突变相对少见。在此前的工作中,我们发现了少见的R528G突变[30],在甲亢合并HPP家系发现了罕见的F671S突变,但是在后续的散发性PP和甲亢PP患者以及5个家系研究中未再有阳性突变发现。国内其他学者也遇到过类似阳性率低的现象,甚至有突变阴性的报道。柯青等[25]在14个家系中对先证者CACNA1S、SCN4A基因突变热点进行了筛查,结果仅发现1例R1239H突变及2例R672H突变,基因突变阳性率为21.4%;随后对71例散发患者进行测序,仅1例存在R672C突变。台湾学者筛查了甲亢和散发性PP患者CACNA1S和SCN4A基因常见的突变R528H、R1239H,结果36例甲亢PP患者突变阴性,12例散发性PP患者仅1例存在R528H突变,散发性PP的阳性率为8.3%[23]。此后台湾学者Sung等[31]报道,60例散发性PP患者在CACNA1S及SCN4A基因中只发现了4例突变,包括1例R1239H、2例R669H和1例R1135H,阳性率仅6.6%。与此相类似,Kung等[32-33]也曾分别对97例和49例甲亢PP的中国香港人群进行基因测序,均未发现突变。
根据目前已发表的大量文献,中国人群中CACNA1S基因和SCN4A基因的突变阳性率远低于西方白种人,提示HPP的突变基因存在种族差异;除了目前报道的突变基因外,中国人群还存在其他致病基因或导致血钾降低的因素。虽然在西方白种人中,HPP的发生原因是CACNA1S和SCN4A基因突变,但中国人群的发病原因可能与西方人群并不完全一致。因此,在此后的研究中,我们考虑需要寻找与西方白种人不同的突变基因,不能只局限于CACNA1S基因和SCN4A基因;同时可通过建立已知常见突变位点的动物模型,对其进行病理生理研究,以促进基因突变的研究。
[1] Peng CY, Pu CQ. Clinical significance of increase of serum creatine phosphate kinase in patients with periodic paralysis[J].Med J Chin PLA, 1999, 24(2)∶ 65-66. [彭超英, 蒲传强. 周期性麻痹患者血清肌酸磷酸激酶增高的临床意义[J]. 解放军医学杂志, 1999, 24(2)∶ 65-66.]
[2] Qi KL. Clinical analysis in periodic paralysis with 57 cases[J].Acta Acad Med CPAF, 2011, 20(8)∶ 650-651.[戚克岭. 周期性麻痹57例临床分析[J]. 武警医学院学报, 2011, 20(8)∶ 650-651.]
[3] Xin SM, Zhang QF, Xin MY. Clinical analysis of hyperthyreosis Complicated hypokalemic periodic paralysis[J]. Chin J Pract Intern Med, 2001, 21(6)∶ 353-354.[辛世萌, 张其芳, 辛敏义.甲亢合并周期性麻痹21例临床分析[J]. 中国实用内科杂志,2001, 21(6)∶ 353-354.]
[4] Ptácek LJ, Tawil R, Griggs RC, et al. Dihydropyridine receptor mutations cause hypokalemic periodic paralysis[J]. Cell, 1994,77(6)∶ 863-868.
[5] Jurkat-Rott K, Lehmann-Horn F, Elbaz A, et al. A calcium channel mutation causing hypokalemic periodic paralysis[J].Hum Mol Genet, 1994, 3(8)∶ 1415-1419.
[6] Bulman DE, Scoggan KA, van Oene MD, et al. A novel sodium channel mutation in a family with hypokalemic periodic paralysis[J]. Neurology, 1999, 53(9)∶ 1932-1936.
[7] Jia JP, Cui LY, Wang W, et al. Neurology[M]. 6th ed. Beijing∶People's Medical Publishing House, 2008. 366. [贾建平, 崔丽英, 王伟, 等. 神经病学[M]. 第6版. 北京∶ 人民卫生出版社,2008. 366. ]
[8] Chabrier S, Monnier N, Lunardi J. Early onset of hypokalaemic periodic paralysis caused by a novel mutation of the CACNA1S gene[J]. Med Genet, 2008, 45(10)∶ 686-688.
[9] Winczewska-Wiktor A, Steinborn B, Lehman-Horn F, et al.Myopathy as the first symptom of hypokalemic periodic paralysis-case report of a girl from a Polish family with CACNA1S (R1239G) mutation[J]. Adv Med Sci, 2007,52(Suppl 1)∶ 155-157.
[10] Meyer T, Jurkat-Rott K, Huebner A, et al. Progressive muscle atrophy with hypokalemic periodic paralysis and calcium channel mutation[J]. Muscle Nerve, 2008, 37(1)∶ 120-124.
[11] Kusumi M, Kumada H, Adachi Y, et al. Muscle weakness in a Japanese family of Arg1239His mutation hypokalemic periodic paralysis[J]. Psychiatry Clin Neurosci, 2001, 55(5)∶ 539 - 541.
[12] Kim JB, Lee KY, Hur JK. A Korean family of hypokalemic periodic paralysis with mutation in a voltage-gated calcium channel (R1239G) [J]. J Korean Med Sci, 2005, 20(1)∶ 162-165.
[13] Ke Q, Xu QG, Huang DH, et al. The mutation R672H in SCN4A gene exists in Chinese patients with hypokalaemic periodic paralysis[J]. Zhonghua Yi Xue Za Zhi, 2006, 86(11)∶ 724-727.
[14] Ke Q, Wu WP, Guo XH, et al. R1239H mutation of CACNA1S gene in a Chinese family with hypokalaemic periodic paralysis[J]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi, 2006, 23 (3)∶ 272-274.
[15] Kageyama K, Terui K, Tsutaya S, et al. Gene analysis of the calcium channel 1 subunit and clinical studies for two patients with hypokalemic periodic paralysis[J]. J Endocrinol Invest,2006, 29(10)∶ 928-933.
[16] Kil TH, Kim JB. Severe respiratory phenotype caused by a de novo Arg528Gly mutation in the CACNA1S gene in a patient with hypokalemic periodic paralysis[J]. Eur J Paediatr Neurol,2010, 14(3)∶ 278-281.
[17] Hirano M, Kokunai Y, Nagai A, et al. A novel mutation in the calcium channel gene in a family with hypokalemic periodic paralysis[J]. J Neurol Sci, 2011, 309(1-2)∶ 9-11.
[18] Kim H, Hwang H, Cheong HI,et al.Hypokalemic periodic paralysis; two different genes responsible for similar clinical manifestations[J]. Korean J Pediatr, 2011, 54(11)∶ 473-476.
[19] Li FF, Li QQ, Tan ZX, et al. A novel mutation in CACNA1S gene associated with hypokalemic periodic paralysis which has a gender difference in the penetrance[J]. J Mol Neurosci, 2012,46(2)∶ 378-383.
[20] Kim JB, Kim MH, Lee SJ,et al. The genotype and clinical phenotype of Korean patients with familial hypokalemic periodic paralysis[J]. J Korean Med Sci, 2007, 22(6)∶ 946-951.
[21] Wang W, Jiang L, Ye L, et al. Mutation screening in Chinese hypokalemic periodic paralysis patients[J]. Mol Genet Metab,2006, 87(4)∶ 359-363.
[22] Wang Q, Liu M, Xu C, et al. Novel CACNA1S mutation causes autosomal dominant hypokalemic periodic paralysis in a Chinese family[J]. J Mol Med (Berl), 2005, 83(3)∶ 203-208.
[23] Lin SH, Hsu YD, Cheng NL, et al. Skeletal muscle dihydropyridine-sensitive calcium channel (CACNA1S) gene mutations in chinese patients with hypokalemic periodic paralysis[J]. Am J Med Sci, 2005, 329(2)∶ 66-70.
[24] Kim SH, Kim UK, Chae JJ, et al. Identification of mutations including de novo mutations in Korean patients with hypokalaemic periodic paralysis[J]. Nephrol Dial Transplant,2001, 16(5)∶ 939-944.
[25] Ke Q, Wu WP, Xu JG, et al. Correlating phenotype and genotype in the familial hypokalaemic periodic paralysis[J]. Chin J Neuro,2006, 39(5)∶ 323-327.[柯青, 吴卫平, 徐金刚, 等. 家族性低钾型周期性麻痹的基因突变与临床特征[J]. 中华神经科杂志,2006, 39(5)∶ 323-327.]
[26] Peng CY, Pu CQ. Clinical analysis of 59 case of graves disease with periodic paralysis[J].Med J Chin PLA, 2003, 28(7)∶ 648-649.[彭超英, 蒲传强. Graves病合并周期性麻痹59例分析[J].解放军医学杂志, 2003, 28(7)∶ 648-649.]
[27] Yang K. Analysis on hyperthyroidism complicated with periodic paralysis in 35 cases[J]. Acta Acad Med CPAF, 2011, 20(8)∶650-651. [杨魁. 甲亢合并周期性麻痹35例临床分析[J]. 武警医学院学报, 2011, 20(8)∶ 650-651.]
[28] Sternberg D, Maisonobe T, Jurkat-Rott K, et al. Hypokalaemic periodic paralysis type2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A[J]. Brain, 2001, 124(6)∶1091-1099.
[29] Matthews E, Labrum R, Sweeney MG, et al. Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis[J]. Neurology, 2009, 72(18)∶ 1544-1547.
[30] Yao HB, Wang XY, Yin CY, et al. A New CACNL1 A3 Mutation in a Chinese Family with Hypokalemic Familial Periodic Paralysis∶Arg528Gly[J]. J Nav Gen Hosp PLA, 2005, 18(2)∶ 65-69.[姚合斌, 王晓英, 尹义存, 等. 家庭性低血钾型周期性麻痹CACNL1A3基因新的突变点528精氨酸被甘氨酸替代[J].海军总医院学报, 2005, 18(2)∶ 65-69.]
[31] Sung CC, Cheng CJ, Lo YF, et al. Genotype and phenotype analysis of patients with sporadic periodic paralysis[J]. Am J Med Sci, 2012, 343(4)∶ 281-285.
[32] Kung AW, Lau KS, Fong GC, et al. Association of novel single nucleotide polymorphisms in the calcium channel alpha 1 subunit gene (Ca(v)1.1) and thyrotoxic periodic paralysis[J]. J Clin Endocrinol Metab, 2004, 89(3)∶ 1340-1345.
[33] Kung AW, Lau KS, Cheung WM, et al. Thyrotoxic periodic paralysis and polymorphisms of sodium-potassium ATPase genes[J]. Clin Endocrinol (Oxf), 2006, 64(2)∶ 158-161.
