Lewis-Sumner综合征6例报告与文献复习
2012-11-23周世梅张在强
周世梅 张在强
Lewis-Sumner综合征(Lewis-Sumner syndrome,LSS),即多灶性获得性脱髓鞘性感觉运动神经病(multifocal acquired demyelinating sensory and motor neuropathy,MADSAM),由Lewis及其同事于 1982年首先报道[1],表现为慢性感觉运动性多发性单神经病,通常以单侧上肢起病,远端受累为主,呈多灶性、不对称分布,电生理学检查提示存在运动神经传导阻滞(conduction block,CB)[1-2]。 LSS 与典型的慢性炎症性脱髓鞘性多发性神经根神经病(chronic inflammatory demyelinating polyneuropathy,CIDP)及多灶性运动神经病(multifocal motor neuropathy,MMN)有许多相似之处,但又有本身鲜明的特点[2],对免疫调节治疗的疗效也不尽相同[3-4],目前倾向于将其视为 CIDP的一种亚型[2-3]。从治疗的角度来讲,将LSS在诸多慢性免疫介导性神经病中准确定位具有重要意义。我们回顾性收集6例LSS患者的临床资料,归纳其主要临床特点,随访其病程演变和治疗反应情况,目的在于加强对该病的认识,减少临床误诊和漏诊,以指导规范化治疗。
1 对象与方法
1.1 研究对象 回顾性收集2007年10月至2010年10月北京天坛医院神经内科确诊的6例Lewis⁃Sumner综合征患者的临床资料。入组标准:①病程≥2个月;②临床表现为不对称性感觉或感觉运动性神经病;③电生理学检查符合2010年欧洲神经病学联盟/周围神经学会(European Federation of Neurological Societies/Peripheral Nerve Society,EFNS/PNS)制定的 CIDP 诊断标准[4]。 排除标准:①系统性血管炎;②单克隆丙种球蛋白病;③副肿瘤综合征;④感染性周围神经病;⑤遗传性神经病。
1.2 方法
1.2.1 临床表现 收集患者病历资料,包括性别、起病年龄、病程、首发症状、症状体征及其分布特点以及治疗、预后等。
1.2.2 辅助检查 收集所有患者实验室检查结果,包括脑脊液细胞计数、蛋白浓度;血清/脑脊液抗 MAG(髓鞘相关蛋白)、GM1、GQ1b、GD1b(神经节苷脂)抗体;脑脊液 /血清 Ho、Yo、Ri抗体;血清及尿液免疫固定电泳;血清抗核抗体、抗中性粒细胞胞浆抗体、抗磷脂抗体、血沉、抗链球菌溶血O及类风湿因子等免疫学检查。收集颈腰椎核磁共振检查结果。部分患者行腓肠神经或腓肠神经-皮肤联合活检。
1.2.3 神经电生理学 所有患者行常规肌电图及节段性传导检查,所检神经有正中神经、尺神经、腓神经、胫神经及腓肠神经,检查项目包括运动神经传导检测、感觉神经传导检测、H-反射、F波,主要记录参数包括远端运动潜伏期、运动神经传导速度、复合肌肉动作电位波幅、运动神经传导阻滞(conduction block,CB)、波形离散(temporal dispersion,TD)、F波出现率及F波平均潜伏期、感觉神经传导速度、感觉神经动作电位波幅。
1.2.4 治疗及随访 所有患者住院期间均给予甲基强的松龙1000 mg静脉滴注冲击治疗,之后改为强的松口服滴定减量。对所有患者行电话随访,内容包括修正的Rankin评分(mRS)、复发情况。
2 结果
2.1 临床特点 6例LSS患者,男、女各3例,发病年龄从24岁到60岁,平均发病年龄为(41±7)岁。从发病到确诊的时间为两个半月到96个月不等,平均病程为45个月。以单侧上肢远端发病者2例,其中1例感觉运动神经纤维均受累,另1例仅累及感觉神经纤维。以单侧下肢远端发病者1例,表现为行走拖曳。另外3例患者分别以四肢近端无力、四肢远端麻木无力及单眼视力下降发病。所有患者就诊时均有感觉、运动神经纤维受累的症状体征,肌无力6例,肌肉萎缩4例,其中2例患者存在肌束颤动,所有患者均存在腱反射减低;有踏棉感者2例,查体音叉振动觉减退者5例,存在肢体麻木者5例,有疼痛症状者1例,所有患者均存在针刺觉减退。就诊时所有患者均有同侧上肢(以下肢发病者)或下肢(以上肢发病者)及对侧肢体受累,1例患者只累及四肢远端,其余患者均存在不同程度近端肢体受累。有3例患者存在颅神经受累的表现,其中1例有右侧面神经炎病史13年,瞬目反射检查示三叉神经传导通路障碍;1例查体有第Ⅴ对颅神经及第Ⅶ对颅神经受累的表现;1例以视物不清发病,脑干听觉诱发电位检查示双侧外周段传导通路障碍。
2.2 实验室、影像学、病理学检查 脑脊液检查显示所有患者白细胞数均 <5/μL,2例脑脊液蛋白含量正常,其余4例均增高,平均蛋白含量为111.5 mg/dL(24.0~ 233.0 mg/dL)。5例行脑脊液/血抗 MAG、GM1、GQ1b、GD1b 抗体检查,除 1 例血清抗GM1⁃IgM(+)外,其余患者均为阴性。脑脊液/血清Ho、Yo、Ri抗体检查,血清及尿蛋白电泳检查及免疫学相关检查均未发现异常。4例行颈椎/腰椎MRI检查显示颈神经根和臂丛神经增粗,注药后强化(图1~3)。4例患者行神经或神经-皮肤活检,存在髓鞘肿胀、脱髓鞘改变,神经内膜少许炎性细胞浸润,未见明显洋葱球样改变,部分神经可见髓球形成。
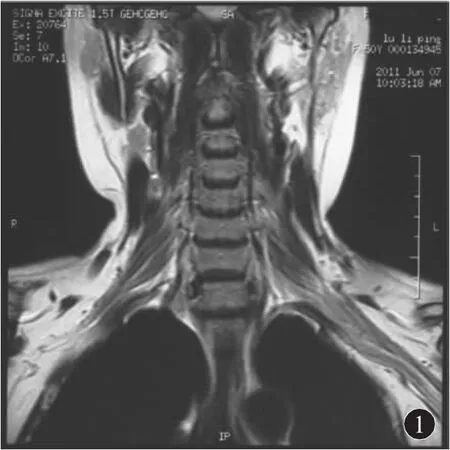
图1 冠状位T1加权相,见双侧颈神经根增粗,呈长T1信号
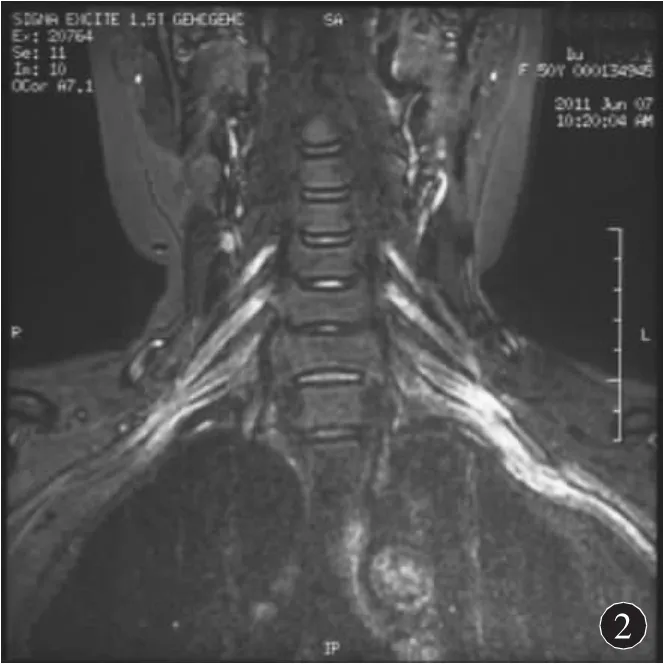
图2 冠状位T1加权钆增强相,见双侧颈神经根增粗并强化
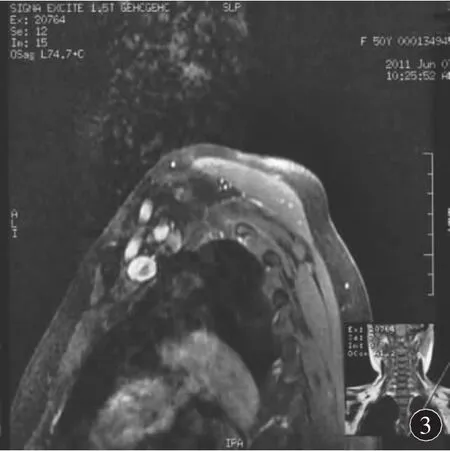
图3 轴位T1加权增强扫描相,见右侧颈神经根增粗,明显强化
2.3 电生理检查 6例患者电生理检查均符合EFNS/PNS制定的“确定的”CIDP电生理改变诊断标准[4]。5例行节段性神经传导检测,均存在传导阻滞,3例可检测到波形离散。所有患者均有远端运动潜伏期延长及运动神经传导速度减慢。所检神经远端复合肌肉动作电位波幅均有下降,但1例患者仅双侧腓总神经远端复合肌肉动作电位波幅下降,双侧尺神经、胫神经及右侧正中神经存在传导阻滞但远端复合肌肉动作电位波幅未见减低。5例患者行F波检查,其中4例存在F波异常(波形未引出、出现率减低和潜伏期延长);2例患者行H反射检查,均未引出波形。除1例外,所有患者均存在感觉神经动作电位波形未引出或感觉神经传导速度减慢。
2.4 治疗及随访结 6例患者均给予激素治疗,临床症状均有一定程度好转,其中4例患者出院时肌力较入院时改善1个级别。6例患者中有2例失访,对4例患者进行电话随访(随访期12~42个月,平均26个月),3例患者病情稳定,可以生活自理、正常上班,但仍有不同程度的遗留症状,mRS评分为1分;1例患者出院后1月自行停用强的松,出现症状复发加重,随访时mRS评分2分。
3 讨论
3.1 临床特点 LSS属于CIDP的不典型亚型,其患病率难以准确估计,Viala与David报道其患病率分别为典型 CIDP 的 1/5[3]和 7 ~ 15%[6],英国最新的流行病学数据显示 LSS占CIDP的15.2%[7]。本研究男性、女性患者病例数相同,而文献报道男性患者多见[3,7],考虑与入组病例数较少有关。文献报道平均发病年龄为40~50岁[3],本组患者为41岁。6例患者均表现为隐袭起病,慢性进展,平均病程为45个月,与国外文献报道相比(24个月)病程偏长[3],考虑可能与以下原因有关:目前尚没有明确的LSS诊断标准,临床大夫对该病认识不足,易误诊为CIDP或MMN混淆;其次,因其以单侧上肢起病多见,常累及正中神经及尺神经,容易误诊为嵌压性神经病;此外,并非所有患者均常规行节段性传导检测,尤其近端节段,难以发现传导阻滞的证据,增加了诊断的难度。文献报道LSS以单侧上肢起病者多见[7],我们的病例中有 2例(34%)患者以单侧上肢起病,1例(17%)以单侧下肢起病,1例(17%)患者以单侧视神经受累起病。本组病例中以感觉运动症状、单纯运动症状、单纯感觉症状发病者分别占三分之一,文献报道首发症状以感觉运动神经纤维受累多见[3]。病程中3例(50%)患者出现颅神经损害,高于文献报道的26%[3],累及视神经、三叉神经、面神经及位听神经,本组病例中没有见到文献所报道的动眼神经受累。有1例(17%)存在疼痛,而Lo YL等[9]报告3例亚洲地区LSS患者均存在疼痛。4例(66%)存在远端肌肉萎缩,5例(83%)患者就诊时即存在近端肌无力,高于文献报道(约22%)[3],但肢体无力仍以远端为著。所有患者就诊时均有四肢受累的表现,3例以单肢起病的患者均进展至其余肢体,而文献报道约50%的LSS患者疾病进展多年后仍保持多灶性分布的特点,较起病之初无明显变化;另50%以单肢起病的患者逐渐进展累及其余肢体,感觉症状分布趋于对称[3]。有文献报道复发型的病例高达66%,亦有文献报道进展型占90%,我们的6例患者中有2例(34%)病程中有缓解,其余患者均为进展性病程。
3.2 辅助检查 与大多数报告一致,我们的病例中有 5例(84%)血清/脑脊液IgM-抗GM1抗体为阴性,1例血清IgM-抗GM1抗体阳性,但滴度水平较低。此前有学者认为可以根据抗GM1抗体的有无来鉴别MMN与LSS,阳性结果可以排除LSS,但进一步研究表明GM1抗体对MMN的特异性并非100%[7]。有研究发现 75例LSS患者中有 3例(4%)存在轻度抗GM1抗体增高,但没有高滴度(≥1∶6400)的患者。 Yang YW[10]亦有类似报告。 同样的,脑脊液蛋白水平对鉴别MMN与LSS帮助也不大,两种情况下均可见蛋白正常或轻度增高。本组病例中4例(66%)脑脊液蛋白含量增高,其中3例(50%)超过100.0 mg/dL,平均蛋白含量为11.5 mg/dL,而多数文献报告LSS患者脑脊液蛋白多正常或仅轻度增高,Rajabally等[7]报告仅42.9%的患者脑脊液蛋白增高,平均为59.0 mg/dL。分析本组患者脑脊液蛋白含量偏高的原因如下:有研究发现非单侧上肢起病的LSS患者,更容易进展累及其它肢体,脑脊液蛋白水平较单侧上肢起病者偏高[7],本组病例中有仅有2例以单侧上肢起病,其余4例以非单侧上肢起病的患者中有3例脑脊液蛋白含量明显增高;另有研究发现LSS脱髓鞘改变呈局灶性或多灶性累及运动神经中段,神经近端相对保留,电生理检查发现仅22%的患者仅存在近端传导阻滞,单独的F波异常少见[3],但本组病病例中有5例(84%)出现F波波形未引出或出现率减低,提示本组病例神经近端受累显著,可能为蛋白增高的原因之一。
4例患者行颈椎核磁检查显示双侧神经根肥大增粗,文献中亦有相似报告[6]。4例患者行腓肠神经活检,提示存在髓鞘肿胀、脱髓鞘改变,1例存在神经轴索变性。多数文献报道,LSS患者感觉神经活检可见大直径有髓神经纤维髓鞘变薄,呈斑片状脱髓鞘改变,以及与洋葱球样结构相似的改变;撕单纤维处理后可见节段性脱髓鞘及髓鞘再生[2]。本组患者未见洋葱球样结构,该结构是长期慢性脱髓鞘与髓鞘再生的结果,多见于经典型CIDP及腓骨肌萎缩症 1型[11]。 与MMN相似,LSS呈多灶性脱髓鞘改变[12],局部腓肠神经活检阳性检出率低,且LSS以CB为特征性电生理改变,研究认为CB的发生机制与郎飞氏节及节旁离子通道改变有关,而非均为脱髓鞘改变[13]。
3.3 神经电生理学 传导阻滞是本组患者电生理学检查的显著特点。上肢及下肢均可见CB,而文献报道CB以上肢多见,即使在下肢受累为著的患者情况亦是如此[3],原因可能为本组病例较国外报道病例有更长的病程,就诊时双侧肢体及上下肢均受累有关。文献报道LSS患者脱髓鞘改变相对避开神经末端,近端神经节段性脱髓鞘也相对少见,电生理学检查发现远端运动潜伏期延长或单独的F波异常并不常见[3]。然而本组病例呈现不同的特点,所有患者均存在远端复合肌肉动作电位波幅减低,80%(4/5,1例未行F波检查)的患者出现F波异常,2例行H反射检查的患者均存在异常,提示本组患者近端神经根及运动神经末梢均有受累。 与多数文献报道一致[5,7],5 例(84%)患者存在感觉神经传导速度减慢、感觉神经动作电位波幅减低或未引出。
3.4 治疗及预后 目前尚没有随机、双盲、安慰剂对照的临床试验进行IVIG与激素或安慰剂疗效比较的研究,但越来越多的证据表明LSS患者可以从IVIG和激素治疗中获益。Benson H等[5]推荐LSS的一线治疗方案为IVIG,预计80%的患者可以从中获益;对IVIG治疗无效的患者,给予强的松口服,激素作为二线用药,预计25%~30%的患者可以获益。另有报告对IVIG和激素治疗无效的顽固性患者,血浆置换可以使病情得到戏剧性改善[14]。本组患者均给予甲基强的松龙1000 mg静脉滴注冲击治疗,1例同时给予环磷酰胺静点后滴定减量,所有患者临床症状均有不同程度改善,其中4例出院时肌力改善1个级别以上。关于本病的预后,文献报告大多数治疗有效的患者需要长期用药以控制复发。Viala等[3]对23例LSS患者平均随访4年发现,26%存在自发缓解稳定期,40%需要长期依赖药物治疗控制症状,10%对治疗无反应。我们随访到4例患者,3例(50%)患者能正常生活、工作,病情稳定,1例(17%)停药后症状复发,目前仍口服激素治疗。
总之,LSS是一种免疫介导性周围神经病,表现为慢性感觉运动性多发性单神经病,通常上肢最先受累,然后进展到下肢,部分病例进展累及对侧肢体,颅神经可以受累;电生理学检查的显著特征为持续存在的传导阻滞,感觉神经动作电位波幅降低;脑脊液蛋白正常或轻度增高。目前倾向于将其视为CIDP的一种变异型,若要确立LSS为一种独立的疾病实体,则需要进一步提供二者存在不同的病理学机制的证据。LSS的一线治疗方案为IVIG 2 g/kg,分 3~5 d给药,每月 1次,共 2~3个月;二线治疗方案为强的松 1 mg/(kg·d),共 4 ~ 6周,然后滴定减量[4];顽固性LSS患者可以试行血浆置换治疗[14]。
[1]Lewis RA,Sumner AJ,Brown MJ,et al.Multifocal demyeli⁃nating neuropathy with persistent conduction block[J].Neurol⁃ogy,1982,32(9):958-964.
[2]Saperstein DS,Katz JS,Amato AA,et al.Clinical spectrum of chronic acquired demyelinating polyneuropathies[J].Muscle Ne⁃rve,2001,24(3):311.
[3]Viala K,Renié L,Maisonobe T,et al.Follow⁃up study and response to treatment in 23 patients with Lewis⁃Sumner syn⁃drome[J].Brain,2004,127(Pt 9):2010-2017.
[4]Van den Bergh PY,Hadden RD,Bouche P,et al.European Federation of Neurological Societies;Peripheral Nerve Society.European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of chronic inflammatory de⁃myelinating polyradiculoneuropathy:report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society⁃first revision[J].Eur J Neurol,2010,17(3):356-363.
[5]Sederholm BH. Treatment of chronicimmune⁃mediated neu⁃ropathies:chronic inflammatory demyelinating polyradiculoneu⁃ropathy,multifocal motor neuropathy,and the Lewis⁃Sumner syndrome[J].Semin Neurol,2010,30(4):443-456.
[6]Allen DC,Smallman CA,Mills KR.Multifocal acquired de⁃myelinating sensory and motor neuropathy presenting as a pe⁃ripheral nerve tumor[J].Muscle Nerve,2006,34(3):373-379.
[7]Rajabally YA,Simpson BS,Beri S,et al.Epidemiologic vari⁃ability of chronic inflammatory demyelinating polyneuropathy with different diagnostic criteria: study of a UK population[J].Muscle Nerve,2009,39(4):432-438.
[8]Rajabally YA,Chavada G.Lewis⁃sumner syndrome of pure up⁃per⁃limb onset: diagnostic,prognostic,and therapeutic fea⁃tures[J].Muscle Nerve,2009,39(2):206-220.
[9]Lo YL,Dan YF,Tan YE,et al.Motor root conduction block in the Lewis⁃Sumner syndrome[J].J ClinNeuromuscul Dis,2011,12(3):158-162.
[10]Yang YW,Liu CH,Tsai CH,et al.Multifocal acquired de⁃myelinating sensory and motor neuropathy:report of a case and review of the literature[J].ActaNeurol Taiwan,2004,13(1):24-28.
[11]梁银杏,葛辉,廖松洁,等.遗传性运动感觉性周围神经病型的临床及电生理特点[J].中国神经精神疾病杂志,2010,36(4):217-219.
[12]Ramchandren S,Lewis RA.Chronic neuropathies⁃chronic in⁃flammatory demyelinating neuropathy and its variants[J].Front Neurol Neurosci,2009,26(6):12-25.
[13]Attarian S,Verschueren A,Franques J,et al.Response to treatment in patients with Lewis⁃Sumner syndrome[J].Muscle Nerve,2011,44(2):179-184.
[14]Park YE,Yook JW,Kim DS.A case of Lewis⁃Sumner syn⁃drome showing dramatic improvement after plasma exchange[J].J Korean Med Sci,2010,25(7):1101-1104.
