新型188RE(Ⅴ)-N-TDD类配合物的结构性质关系研究
2012-05-16魏洪源王关全王文进罗顺忠
魏洪源,王关全,王文进,罗顺忠
(中国工程物理研究院 核物理与化学研究所,四川 绵阳 621900)
186Re、188Re因其具有优良的核素性质,一直是放射性治疗药物研究的热点[1],目前研究最多的是铼的五价化合物,大多数配合物含有以[188Re=O]3+为中心的金属核。受同族[99Tcm≡N]2+核配合物研究结果的启发[2-4],[188Re≡N]2+化合物也得到了更多关注。[188Re≡N]2+化合物具有相应[188Re=O]3+化合物更优的稳定性、不同的电性和脂溶性,因此可能获得更具有应用价值的化合物。本研究小组已合成了S2N2类配体2,2,9,9-四甲基-4,7-二氮-1,10-二硫癸烷(TDD)衍生物,并制备了相应的188Re(Ⅴ)-O-配合物,用于碘化罂粟油的标记,希望获得有前景的放射性肝动脉灌注栓塞治疗剂,但标记物的产率和稳定性都不太理想[5]。此后,在改进[188Re N]int2+中间体制备方法[6]的基础上,制备了含[188Re≡N]2+核的 TDD 类 配 合 物[7-8]。本工作在该优化制备方法基础上,对配合物的体外稳定性、脂溶性和反相HPLC色谱行为等性质进行了测定。密度泛函理论(DFT)在研究99Tcm、188Re配合物的结构、成键和性质方面已取得较好的结果[9-10]。因此,为进一步深入了解所制备的188Re(Ⅴ)-N-TDD类配合物的性质,本工作拟采用DFT方法对配合物的结构和成键进行计算,并使用极化连续介质模型(Polarizable Continuu m Models,PCM)计算配合物在水中的绝对溶剂化自由能。通过结构和性质的关联讨论该类配合物的结构效应,希望通过构效关系的研究减少药物设计中的盲目性,为改进现有配体分子的结构及设计新的配合物提供理论指导。
1 主要试剂与仪器
FJ-2021型γ免疫计数器:西安核仪器厂;HPLC系统:由 BECK MAN 421 Contr oller,BECK MAN 110B Sol vent Delivery Module,BECKMAN Model 170 Radioistope Detector组成;聚酰胺薄层板:浙江台州市路桥四甲生化塑料厂提供;188W-188Re发生器:中国科学院上海应用物理研究所提供。
丁二酰肼 (SDH)、Sn Cl2·2 H2O:美 国Al drich公司提供;其他试剂均为市售分析纯。2,2,9,9-四 甲 基-4,7-二 氮-1,10-二 硫 癸 烷(TDD,L1)、2,2,9,9-四甲基-4,7-二氮-4-乙撑哌啶-1,10-二硫癸烷(NEPTDD,L2)、2,2,9,9-四甲基-4,7-二氮-4-乙撑-(4-甲基)哌啶-1,10-二硫癸烷(NEMPTDD,L3)和2,2,9,9-四甲基-4,7-二氮-4-乙撑-(3,5-二甲基)哌啶-1,10-二硫癸烷(NEMMPTDD,L4):均为本课题组合成并经过分析确认,其结构简式示于图1。
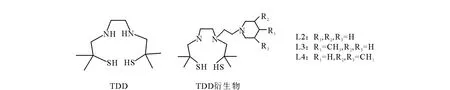
图1 TDD及其衍生物结构简式
2 实验方法
2.1 188 Re N配合物的制备和体外性质测定
2.1.1188Re N配合物的制备
依照本课题组改进的中间体制备方法[7-8]制备[188Re N]int2+中间体。其放化纯度大于98%,放射性浓度约为148 GBq/L。
取0.5 mL [188Re N]1nt2+中间体,分别加入1 mL(10 g·L-1)相应的配体,调p H至7,70℃水浴反应30 min。溶液由棕黄色变为无色,得到相应的188Re N配合物。
使用TLC和反相HPLC测定188Re N配合物的标记率和放化纯度。TLC分析时,以聚酰胺薄膜为支持体,生理盐水和乙腈为展开剂。反相 HPLC分析时,固定相为C-18反相柱(250 mm×4.6 mm,Dia monsil T M),流动相为0.1%三氟乙酸(TFA)水溶液和乙腈混合液梯度淋洗,流速为1 mL·min-1。
2.1.2188Re N配合物的脂水分配系数
采用Shaking Flask法测定188Re N配合物的脂水分配系数(l g PO/W):向10 mL离心管中加入1.4 mL PBS(每100 mL含116 mg Na OH和680 mg KH2PO4,p H=7.4)溶液和0.1 mL制备好的188Re N配合物,再加入1.5 mL经PBS饱和的正辛醇,振摇15 min,3 000 r/min离心15 min。分别取0.1 mL有机相和水相,测定两相放射性计数,计算其lg PO/W。
2.1.3188Re N配合物的体外稳定性
标记原液的稳定性:将制备好的188Re N配合物敝口放置,于不同时间点取样,用反相HPLC分析其放化纯度。
生理盐水中的稳定性:将制备好的配合物用生理盐水稀释10倍和100倍,37℃温育后用反相HPLC测不同时间点的放化纯度。
小牛血清溶液中的稳定性:188Re N配合物加入到1.0 mL小牛血清中(预先用100 mmol/L、p H7.0的磷酸缓冲溶液稀释,体积比为1∶100),37℃温育后于不同时间点取样,在所取样品中加入乙腈后离心,取上清液分析其放化纯度。
2.2 体外竞争实验
取适量配合物分别加入预先配制的1.0和10.0 mmol/L的L-半胱氨酸的磷酸缓冲溶液(100 mmol/L,p H7.0)、组胺酸或0.02 mol/L谷胱甘肽或0.02 mol/L牛血清白蛋白,另以等量的生理盐水作对照。37℃温育,于不同时间点分析放化纯度。
188Re N配合物的电性测定:点样于新华滤纸中部,磷酸盐缓冲液(p H7.4)作溶液介质,电压150 V,电泳90 min。此后测量正、负极及原点的放射性计数,并计算其占总放射性计数的百分比。
2.3 配合物的密度泛函和溶剂化自由能计算
用DFT的B3LYP(Gaussian03程序)方法对配合物进行结构优化及能量计算[9]。C、H、O、N、S原子采用6-31G*基组,金属Re使用双-ζ的LANL2DZ赝势基组。配合物初始结构参考与文献[11-12]相近似的配合物建模,电子布居分析使用Mulliken Population Analysis。使用自洽反应场理论(SCRF)计算配合物的绝对溶剂化自由能,方法为极化连续介质模型(PCM)。
3 结果与讨论
3.1 188 Re N配合物的制备和性质测定
3.1.1188Re N配合物的制备
188Re N配合物的制备为两步法,即先制备高纯度中间体[188Re N]int2+,再通过配体交换反应获得相应188Re N配合物。[188Re N]int2+的制备过程中N给予体、促进剂和还原剂等是决定产率的关键因素。在Tc≡N核配合物研究[2-3]中,曾使用一些制备相对困难的硫代酰肼作为N给予体。本工作选择丁二酰肼(SDH)作为N给予体,产率可达95%以上,且中间体单一。优化的中间体制备条件为:1.0 mL (10 g·L-1)SDH、0.6 mL(100 g·L-1)草酸、0.2 mL188Re O4-(生理盐水淋洗液,约37 GBq·L-1)、0.2 mL(1 g·L-1)NH4Re O4、0.2 mL (10 g·L-1)Sn Cl2·2 H2O,调p H 至2.0,室温反应30 min,得到[Re N]int2+中间体,反应液呈棕黄色。采用HPLC分析所得中间体,结果示于图2。由图2可知,中间体保留时间tR1=16.8 min(图2a),188Re O4-保留时间tR2=19.5 min(图2b)。
[Re N]int2+中间体合成过程中,草酸作为促进剂,其作用非常明显,如在SDH用量为10 mg,NH4Re O4用量为0.2 mg,188Re O4-淋洗液用量为0.2 mL(约37 MBq·mL-1),Sn Cl2·2 H2O用量为2 mg,p H 为2.0,室温反应30 min的条件下,草酸使用量对产率的影响列于表1。由表1可以看出,当草酸用量从15 mg提高到60 mg时,产率也从88.5%上升到96.0%。
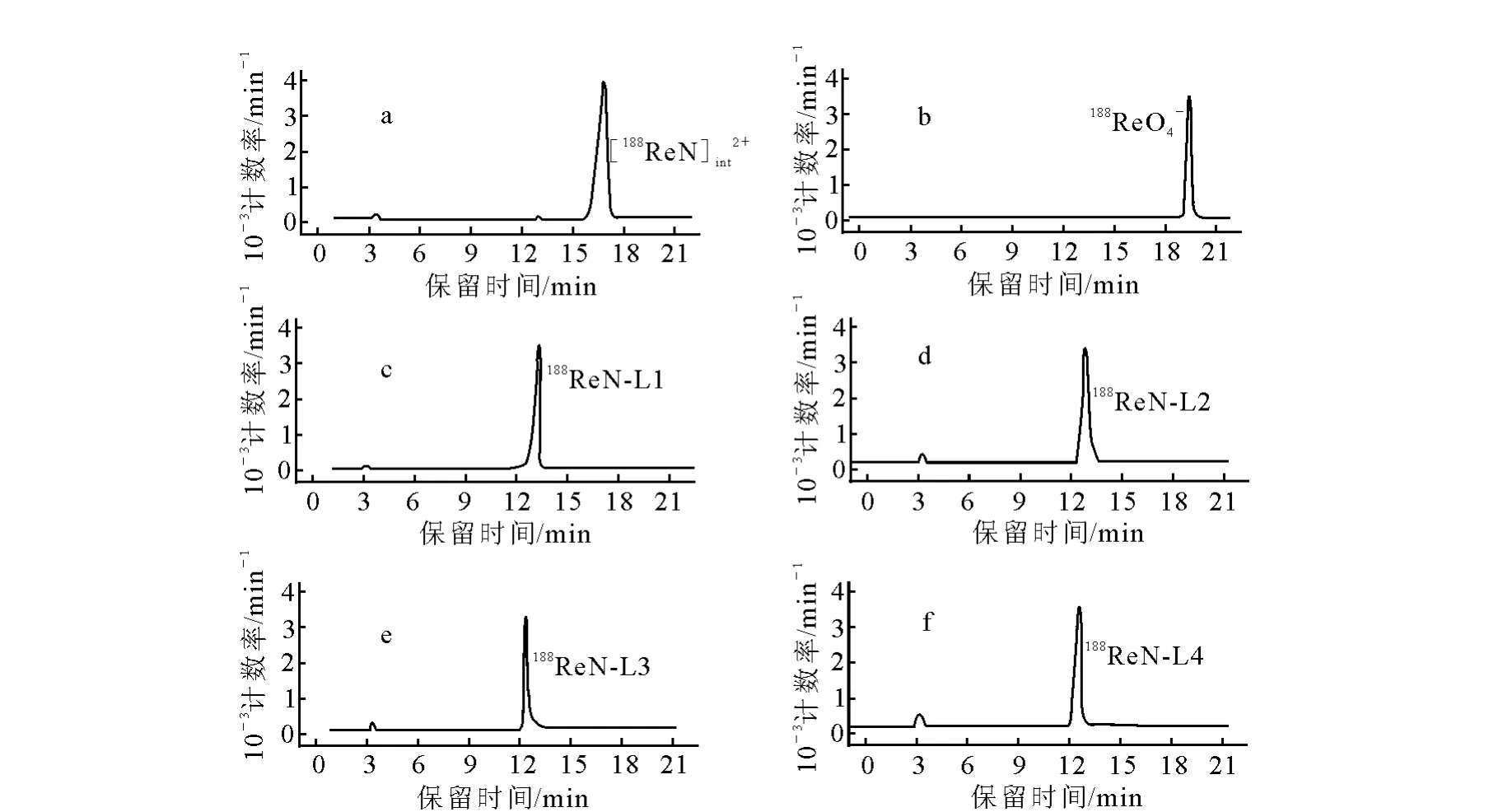
图2 不同配合物体系的HPLC图谱
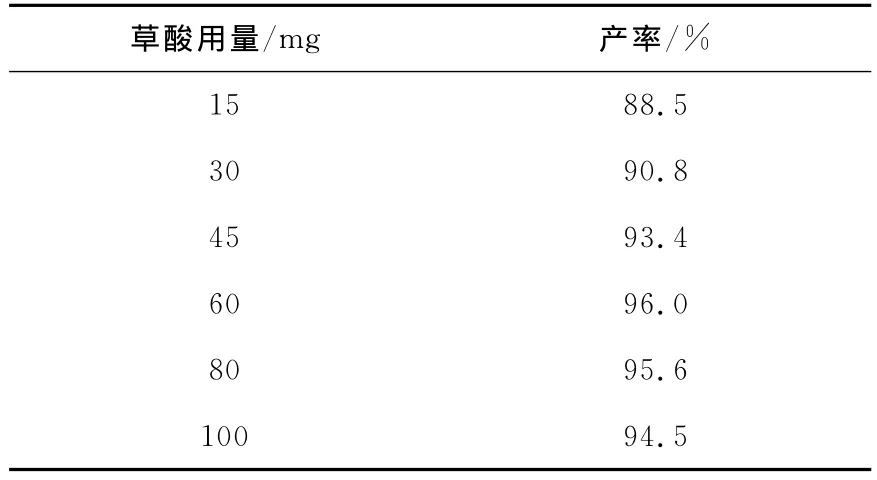
表1 草酸用量对产率的影响
188Re O-还原制备放射性药物时,188Re(Ⅶ)中心必须从四配位、四氧阴离子转变为通常有五到六配位的最终化合物,即分子几何结构从四面体变为一个扩张的四方锥或者八面体结构。一般来说,这个几何结构的变化过程使标准还原电势Eo急剧降低。曾有学者[13]基于配位化学中“配位构型扩张”的理论[14]提出该问题解决方法,即可以通过一些合适的试剂把四面体高铼酸阴离子变为一个四方锥或者八面体188Re(Ⅶ)中间体,但不改变Re氧化状态来达到消除Eo急剧降低的效果。草酸根离子(C2O42-)就是一个可以生成具有扩张的配位几何结构的188Re(Ⅶ)中间体的试剂,能显著提高最终配合物的产率。
在中间体溶液加入相应配体,中性条件下反应30 min即可得到相应的配合物。使用TLC和HPLC分析其产率和放化纯度。HPLC分析中,使用常规的流动相进液顺序(先高极性后低极性)无法将3种配合物分开,因此通过多次实验进行优化,优化后进液顺序与常规的方式有所不同,即流速为1 mL· min-1,流动相为:A=0.1%三氟乙酸(TFA)水溶液,B=乙腈;0~7 min,100%B;7~18 min,20%A,80%B;18~20 min,100%A;20~25 min,100%B。所得Re(Ⅴ)-配合物的HPLC结果示于图2。由图2c、图 2d、图 2e、图 2f可知,188Re N-TDD(188Re N-L1)保留时间为tR3=13.2 min;188Re NNEP-TDD(188Re N-L2)的 保 留 时 间 为tR4=12.9 min;188Re N-NEMP-TDD(188Re N-L3)的保留时间为tR5=12.7 min;188Re N-NEMMPTDD(188Re N-L4)的保留时间为tR6=12.4 min。分析表明,标记体系中的主要放射性成分为188Re(Ⅴ)-N-L 配 合 物,其 产 率 和 放 化 纯 度 大 于95%,几乎没有未还原的188Re O4-存在,只有极微量的胶体铼存在(tR=3.4 min)。TLC分析各物质的Rf列于表2。由表2可知,选择的分析条件能够有效分开各个组份,适于本制备体系的分析测定。TLC分析得到了与HPLC分析相同的结果,表明制备方法简便高效,能够获得高产率的铼氮核配合物。
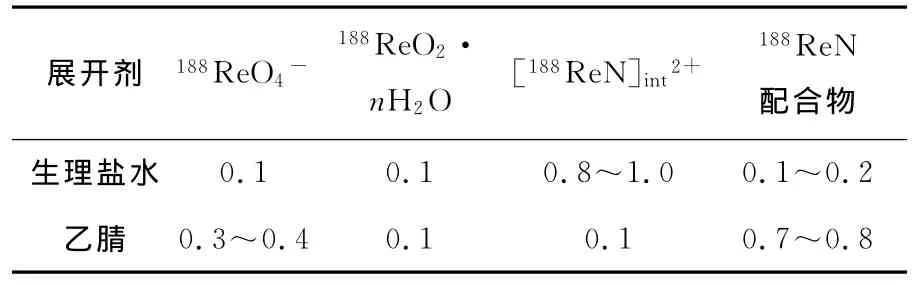
表2 TLC分析中的R f
3.1.2188Re N配合物体外稳定性
室温敞口放置24 h,配合物放化纯度几乎没有降低。稀释后和在小牛血清溶液中温育24 h后,放化纯度仍大于90%。在竞争配体L-半胱氨酸、组氨酸、谷胱甘肽和牛血清白蛋白存在情况下,12 h后的放化纯度变化很小,仍均大于85%,提示配合物有较好的体外稳定性。
3.1.3188Re N配合物脂水分配系数
188Re N-L1、188Re N-L2、188Re N-L3、188Re NL4的脂水分配系数(l g PO/W)分别为1.12、1.43、1.48和1.64,为脂溶性较好的配合物,且其脂溶性 按188Re N-L1<188Re N-L2<188Re N-L3<188Re N-L4顺序增大。
电泳实验结果显示,有97%以上都停留在原点,负极的放射性为2.6%,正极的为0.4%,表明所有188Re N配合物都为电中性物质(零电荷)。此结果进一步证明188Re N配合物有良好的脂溶性。
3.2 配合物的DFT计算和构效关系
配合物的三维结构示意图示于图3。对于本工作涉及的N2S2类四齿配体,与Re直接配位的两个N原子为sp3杂化,其中一个N原子上连接有取代基。根据sp3杂化N原子上取代基R或H原子相对于Re≡N(或Re=O)的空间取向,可能存在syn(两者都位于S2N2近似平面的同侧)和anti(两者位于S2N2平面的异侧)两种异构体[15]。本工作分别计算了两种异构体的能量和结构。从计算结果来看,所有配合物为五配位,结构为变形的四方锥结构。由于空间位阻效应,所有的syn异构体的能量都比anti异构体的低,差值在10~100 kJ/mol范围内,且取代基越大,这一差值越大,因此配合物的稳定构型均为syn异构体,这与绝大多数的Re-N2S2类配合物的X-衍射晶体结果[9]一致。
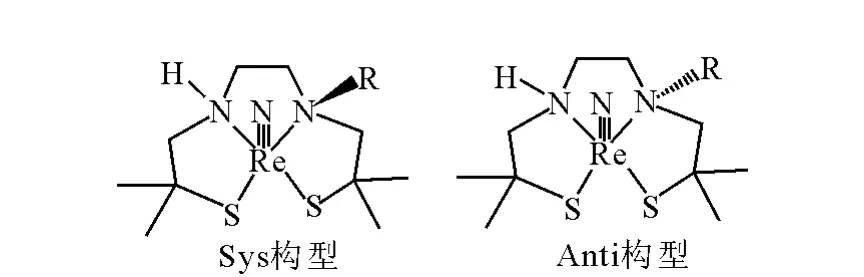
图3 188 Re(Ⅴ)-N-TDD配合物结构简式
本工作对syn构型的主要计算结果列于表3。由表3可知,两个S原子和两个N原子近于同一平面,随着sp3杂化N原子上的取代基R(乙撑哌啶)的加入,该平面有一定的变形,有取代基的N原子略高于平面,二面角S-N-N*-S*也由最初188Re N-TDD 的3.6°增加到188Re NNEPTDD的8.9°。O2-和 N3-都是很强的π授体,可与金属铼形成非常稳定的化学键。与前期制备的该类配体的Re=O核配合物[2]相比,不同铼核配合物中Re≡N键比Re=O[14]的键长短3 p m,Re≡N核上的Re原子的净正电荷比Re=O核上Re原子平均低30%,电子重叠集居数Re≡N比Re=O高约20%。因此N3-是比O2-更强的π授体,Re≡N键比Re=O键更稳定。配体经过乙撑哌啶修饰后,配合物分子的极性明显增加,由于空间位阻增大,使其连接的N原子和Re之间的配位键的键长增长,整个配合物的空间结构的稳定性下降。这一空间位阻效应在Re=O配合物的制备难易程度和稳定性趋势方面体现明显,如188Re-O-L3和188Re-O-L4制备条件更严格,且产率较低(最大约85%),稀释稳定性和竞争稳定性也较差,RCP很快降低到70%。但通过配体交换反应与更强的N3-π授体形成的Re≡N配合物的制备过程中这一现象不明显,反应条件和产率都很近似,产率>95%,且稳定性很好。在乙撑哌啶上增加不同数目的甲基,空间位阻变化几乎为零,不影响与Re≡N核的成键,对结构的影响几乎观察不到,并且分子极性没有变化。
许多物理、化学以及生物过程本质上是由分子之间的相互作用决定,其中疏水作用在药物的代谢、排泄和体内行为中起着重要作用。如本工作希望增大配合物的疏水性(即亲脂性),从而增加铼配合物在碘化罂粟油中溶解性,改善肝动脉灌注栓塞治疗效果。当前疏水作用研究不够成熟,量度困难。由于疏水效应与溶剂分配现象有关,因此通常将分子在油/水体系中的分配系数作为疏水性的宏观度量。一般选择正辛醇-水体系,其原因是该体系更与生物体系接近。分配系数是一种平衡常数,与自由能成对数线性关系(LFER),因此通常以对数形式(l g PO/W)出现在定量构效关系研究中。本工作测定了所制备的Re(Ⅴ)-N-TDD类配合物的l g PO/W,从测量结果来看,亲脂性随着侧链修饰基团的增加而增加,即188Re N-L1<188Re N-L2<188Re N-L3<188Re NL4依次增大,与设计目标一致。化合物在反相HPLC系统上的保留时间和水溶性(或脂溶性)有较好的相关性[16],一般来说,脂性溶性越强,其色谱保留时间越短,因此实验中也常通过建立l g PO/W与色谱保留指数之间的定量关系,再根据较易测定的色谱保留指数来推算l g PO/W,比较其亲水(亲脂)性质,这同样是线性自由能关系的另一种表达形式。不同配合物HPLC保留时间结果表明,保留时间188Re N-L1>188Re N-L2>188Re N-L3>188Re N-L4,即 预 示 着 亲 脂 性 按188Re N-L1<188Re N-L2<188Re N-L3<188Re N-L4顺序增大,与lg PO/W的测定结果一致。
在理论计算方面,自治反应场(SCRF)理论、Onsager模型和PCM模型都能计算溶剂效应对化合物性质的影响[17-19]。其中PCM 模型可以计算化合物的溶剂化自由能,从而定量其亲水(亲脂)性,因此本工作采用了PCM模型计算铼氮核配合物的水溶剂化自由能。在可极化连续势模型(PCM)中,溶剂化自由能定义为完成以下过程所做的可逆功:从溶质分子与溶剂之间相互无作用到在连续分布的溶剂中形成一个空腔并把溶质分子放置于其中。溶剂化后体系总自由能ΔGS由下式计算:
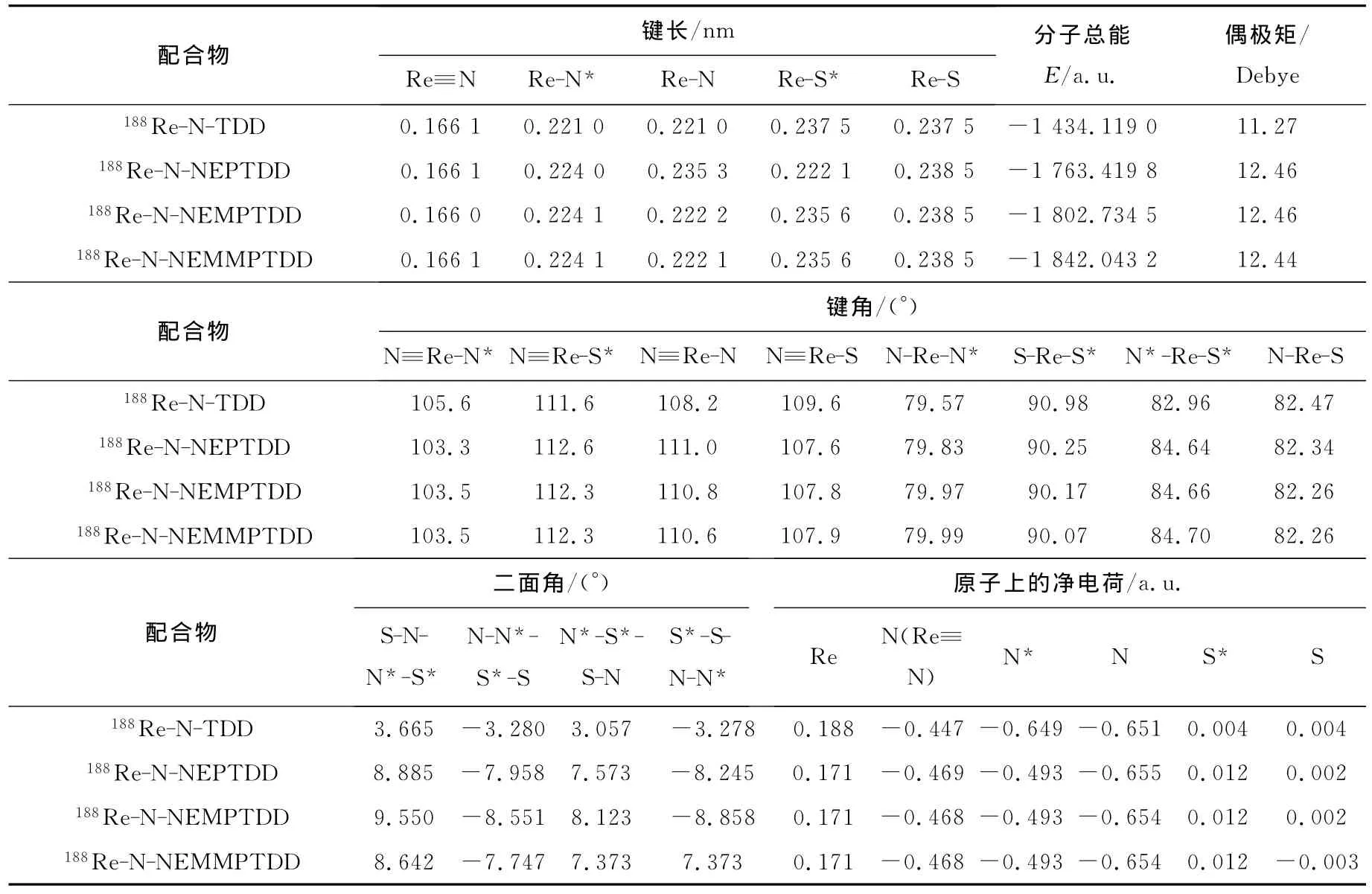
表3 配合物B3LYP计算部分结果
(1)式中,ΔGel为静电能;ΔGnon-elc为非静电能;ΔGdis-rep为溶质-溶剂的色散-排斥能;ΔGcav为空穴能,即在原来应该是溶剂的地方形成一个空腔所需要做的功;ΔGmm包括各种分子运动的贡献,如零点能,振动、平动、转动等等,这一项可以采用经典统计力学方法通过振动分析进行计算。因为在计算时忽略溶剂化前后的分子构型变化,因此Gmm变化常可以忽略。根据此公式计算得188Re(Ⅴ)-N-配合物水溶剂化自由能列于表4。由表4可以看出,配合物的水溶剂化自由能的大小依次为188Re N-L1<188Re N-L2<188Re NL3<188Re N-L4。此结果说明,配合物的水溶性按此顺序减小,而脂溶性按此顺序增大,这与所测定的配合物脂水分配系数的变化趋势一致。从表3中可以看出,静电作用和非静电作用共同决定了配合物的水溶性,当在TDD分子上的N原子引入一个乙撑哌啶基后,分子的极性增加,偶极矩由11.27变化为12.46,这有利于配合物在水中的溶解。但由于增加了一个大的修饰基,使形成空穴的能量增大,从156.86 MJ/mol变为210.26 MJ/mol(37.49~50.22 kcal/mol),从而降低了水溶性。多种贡献的总体决定了188Re-N-NEPTDD的水溶性比188Re-N-TDD的差。继续在哌啶环上增加甲基,对分子的极性几乎没有影响,静电对水溶性的影响不大,同时由于分子体积的增加,使形成空穴所需的能量变大,因此总体上增加了配合物的水溶性。
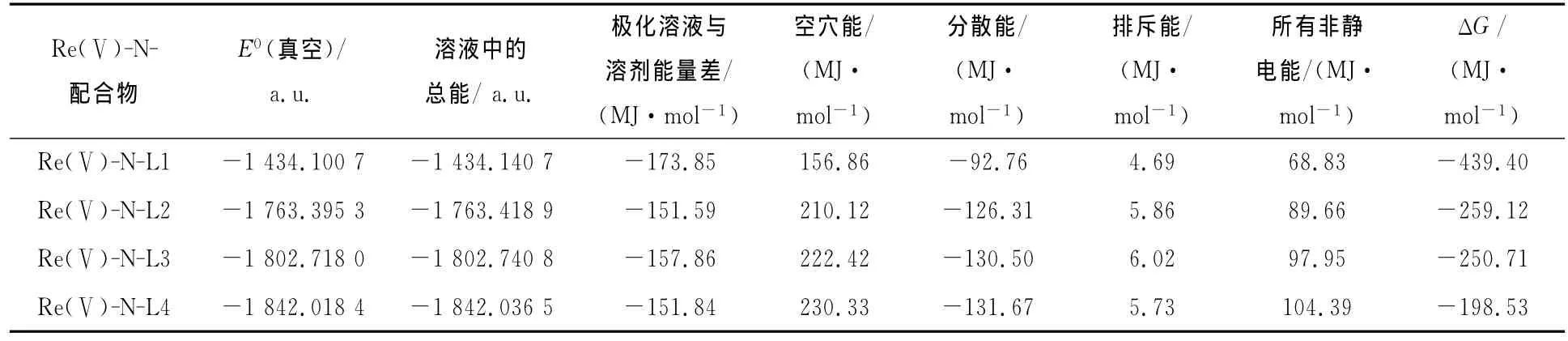
表4 188 Re(Ⅴ)-N-配合物水溶剂化自由能结果
3 结 论
利用改进的[188Re N]int2+中间体制备方法和配体交换法制备了四个188Re(Ⅴ)-N-TDD类配合物,建立了HPLC和TLC分析测定方法,测定了稳定性和脂水分配系数等参数。
B3LYP计算结果能够对此类配合物的结构和成键进行预测。使用PCM模型计算获得了配合物的水溶剂化自由能。构效关系研究表明,溶剂化自由能变化和脂水分配系数有相同的变化趋势,通过预先的理论计算能够对配合物性质定性预测,对该类化合物的设计提供帮助。
[1] Deutsch E,Libson K,Vander heyden JL,et al.The chemistry of rhenium and technetium as related to the use of isotopes of these elements in therapeutic and diagnostic nuclear medicine[J].Int J Rad Appl Instr u m B,1986,13:465-477.
[2] Mangera KO,Vanbilloen HP,Bellande E,et al.Influence of a99Tc N core on the biological and physicochemical behavior of99Tcmco mplexes of L,L-EC and L,L-ECD[J].Nucl Med Biol,1996,23:987-993.
[3] Bolzati C,Boschi A,Uccelli L,et al.Chemistry of the str ong electrophilic metal frag ment[99Tc(N)(PXP)]2+(PXP=diphosphine ligand):A novel tool f or t he selective labeling of s mall molecules[J].J Am Chem Soc,2002,124:11 468-11 479.
[4] Zhang Junbo,Wang Xuebin,Liang Jing.Synthesis of a bis-(N-sec-butyl-dithiocar bamato)-nitrido-99 mTc complex:a potential new radiophar maceutical for brain perf usion studies[J].Applied Radiation and Isotopes,2005,62:33-37.
[5] 王文进.188Re-(烷基)-TDD配合物的制备及性质研究[D].绵阳:中国工程物理研究院,2003.
[6] Boschi A,Bolzati C,Uccelli L,et al.High-yield synt hesis of the ter minal188Re≡N multiple bond from generator-produced[188Re O4]-[J].Nuclear Medicine and Biology,2003,30:381-387.
[7] 王关全,魏洪源,罗顺忠,等.188Re N核配合物制备新方法研究[J].同位素,2008,21(1):25-28.
[8] Wang GQ,Wei HY,Luo SZ,et al.Preparation and pri mary biological evaluation of novel nitrido-188Re co mplex ws/lipiodol[J].Nucl Sci &Tech,2008,19:223-229.
[9] 王东琪,褚泰伟,王祥云,等.[Tc(CO)3(H2O)3]+及其几个衍生物的结构和成键[J].核化学与放射化学,2000,22(4):207-213.
[10]贾红梅,刘伯里,孟昭兴,等.锝化学研究Ⅻ:99Tcm-配合物的溶剂化自由能与其脑吸收值的关系[J].核化学与放射化学,2002,24(1):42-50.
[11]Lee YS,Jeong JM,Ki m YJ,et al.Synthesis of188Re-labeled long chain alkyl diaminedit hiol f or therapy of liver cancer[J].Nucl Med Commun,2002,23:237-242.
[12]Demai may F,Dazord L,Roucoux A,et al.Rhenium-188 and technetiu m-99 m nitridobis(N-ethoxy-N-ethyldithiocar bamate)Leucocyte labeling radiophar maceuticals:[188Re N (NOET)2]and[188Tc N(NOET)2],NOET = Et(Et O)NCS2:t heir in vitro localization and chemical behaviour[J].Nucl Med Biol,1997,24(4):701-705.
[13]Bolzati C,Boschi A,Uccelli L,et al.An alter native appr oach to the preparation of188Re-radiophar maceuticals fr om generator-produced [188Re O4]-:efficient synthesis of188Re(Ⅴ )-DMSA(DMSA-meso-2,3-di mercaptosuccinic acid [J].Nucl Med Biol,2000,27:309-314.
[14]Vajo JJ,Aikens DA,Ashley L,et al.Facile electroreduction of perrhenate in weakly acidic citrate and oxalate media[J].Inor g Chem,1981,20:3 328-3 333.
[15]Jeong JM,Kim YJ,Lee YS,et al.Lipiodol solution of a lipophilic agent,188Re-TDD,for the treat ment of liver cancer[J].Nucl Med Biol,2001,28(1):197-204.
[16]Leo A,Hansch C,Elkins D.Partition coefficients and their uses[J].Chem Rev,1971,71:625.
[17]陈凯先,蒋华良,嵇汝运.计算机辅助药物设计:原理、方法及应用[M].上海:上海科学技术出版社,2000:149-151.
[18]Hubinyi H.The quantitive analysis of structureactivity relationship[C]// Wolff ME.Burger’s medicinal chemistry and drug design vol 1:Principles and practice.5thedition.New Yor k:John Wiley &Sons,1995:497.
[19] 王 祥 云,魏 雄 辉,刘 新 起,等.99TcmO3+和99TcmNO2+的二胺基二硫醇配合物在脑中滞留机制的量子化学研究[J].化学学报,2000,58:1 522-1 528.
