茎环法RT-qPCR定量检测细胞抗猪瘟病毒siRNA的表达
2012-02-09刘帅李江南袁婷杨凡力逄大欣涂长春
刘帅,李江南,袁婷,杨凡力,逄大欣,涂长春
1 吉林大学畜牧兽医学院,吉林 长春 130062
2 军事医学科学院军事兽医研究所,吉林 长春 130122
RNAi作为一种有效的抗病毒工具,已成功应用于抑制人体免疫缺损病毒Ⅰ型 (HIV-1)[1]、SARS冠状病毒 (SARS-CoV)[2]等多种病毒的复制增殖。猪瘟 (Classical swine fever,CSF) 是由猪瘟病毒 (Classical swine fever virus,CSFV) 引起的一种高度接触性传染病,给养猪业带来重大经济损失。CSFV是有囊膜的正链RNA病毒,基因组RNA兼具复制和转录功能,且非结构蛋白基因保守性比较高[3],提供了理想的RNAi靶位点。为应用RNAi技术构建抗猪瘟病毒转基因猪,本研究小组曾针对 CSFV非结构蛋白基因Npro成功设计了2个siRNA——siN1和siN2[4],并构建了相应的siRNA表达载体——pLox-siN1和pLox-siN2[5-6],在细胞水平通过检测病毒的复制和增殖水平,证明了siN1和siN2对CSFV有显著的抑制效果,但是,抗性细胞中siRNA的表达检测却制约了RNAi效果的评价。所以亟需建立一种准确而简便的siRNA定量检测方法,对抗猪瘟病毒转基因猪 siRNA的表达水平及组织表达差异进行有效监测及评价。
目前,已有多种siRNA、miRNA (microRNA)等小RNA的检测方法。Northern blotting[7]仍是小RNA检测的金标准,但其特异性和灵敏度不高。而基于LNA (Locked nucleic acid) 标记探针的新型 Northern blotting[8]虽然显著改善了检测灵敏度和特异性,但仍存在操作步骤繁琐、需样品量大、可检测范围低等缺点,更难于准确定量。然而,多种实时定量PCR方法[9-16]被开发出来定量miRNA的表达水平,常用的包括Poly (A) 加尾法[15]和茎环引物法[16]。前者可同时逆转录细胞内所有的 miRNA,成本低但灵敏度和特异性均不如后者,适合于miRNA的大量筛选工作。后者创新地设计了具有稳定空间结构的茎环引物,比常规线性引物有更高的特异性和灵敏度,并联合特异性MGB探针,使其可精确区分同家族高度同源的miRNA,重要的是仅对成熟miRNA进行检测,而不受其前体 (Pre-miRNA) 干扰。但每种miRNA都需要设计一个对应的茎环引物和MGB探针,成本高且操作相对繁琐,所以适于少数靶基因的定量检测。2009年,Cheng等[17]将茎环法 RT-qPCR成功地应用于人工合成siRNA的检测,并在分子、细胞和动物个体等水平准确定量了未修饰的单链siRNA,3末端有两个突出的双链Silencer®siRNA及LNA修饰的双链Silencer®Select siRNA等3种形式的siRNA,有效地解决了对siRNA传递效率、分布和稳定性等难于评价的问题。
在本研究中,我们建立了检测siN1和siN2两个抗CSFV的特异siRNA表达水平的茎环法RT-qPCR,准确定量检测了抗CSFV的PK-15细胞克隆的siRNA表达水平,特异性强,灵敏度高,可精确至100个拷贝,检测范围宽。可用于RNAi抗病毒效果的定量评价,为未来转基因动物的抗病毒效果评价提供了参考。
1 材料与方法
1.1 材料
1.1.1 质粒和细胞
高效且特异抑制CSFV复制增殖的siRNA表达载体pLox-siN1和pLox-siN2及表达乱序siN1的对照载体pLox-siC均为本实验室构建并保存[6];PK-15猪肾传代细胞系 (85代) 由本室保存;猪胚胎成纤维细胞 (Porcine fetal fibroblasts,PFF) 由吉林大学欧阳红生教授惠赠。
1.1.2 主要试剂和仪器
限制性内切酶Apal Ⅰ购自NEB公司;转染试剂FuGENE®HD购自Roche公司;G418购自Gibco公司;总 RNA 提取试剂盒 (MirVana miRNA Isolation Kit),逆转录试剂盒 (TaqMan®MicroRNA Reverse Transcription Kit) 和实时定量PCR试剂 (TaqMan Universal Master Mix,No AmpErase®UNG) 均购自 ABI公司;猪抗CSFV阳性血清 (5号) 和阴性猪血清由本实验室制备、鉴定并保存[18];异硫氰酸荧光素(FITC) 标记的兔抗猪IgG抗体购自Sigma公司;实时定量 PCR仪 Stratagene Mx3000P购自Agilent公司;核酸测定仪 NanoDrop 1000购自Thermo公司。
1.2 转染及抗CSFV的阳性细胞克隆鉴定
用Apal Ⅰ双酶切质粒pLox-siN1、pLox-siN2和pLox-siC并乙醇沉淀回收含抗性基因neo的目的片段。用含10%小牛血清的MEM营养液以每孔2.0×105个PK-15细胞传代于6孔细胞培养板中,37 ℃、5% CO2培养箱中培养20~24 h,使细胞贴壁生长满度达到85%~90%,用FuGENE®HD转染24 h后1∶50传代于6孔细胞培养板中,培养24 h后换用500 mg/L G418的MEM培养基筛选 12~14 d,质粒 pLox-siN1、pLox-siN2和pLox-siC挑取细胞克隆 (分别命名为 PK-N1、PK-N2和PK-siC) 各3个至24孔板中培养,扩大培养并进行neo基因PCR鉴定 (引物neo-FP和neo-RP序列见表1),以确定基因组中整合了目的基因片段。按以前所述方法[6]包括IFA (接毒后72 h)、病毒基因组拷贝数 (48 h和72 h) 和TCID50(48 h和 72 h) 测定以上阳性克隆抑制CSFV复制增殖的效果。
1.3 引物和MGB探针设计
参考 Chen等[16]和 Tang等[19]设计 siN1和siN2检测用引物和探针 (表 1):茎环引物由 5′端通用茎环结构序列 (5′-CTCAACTGGTGTC GTGGAGTCGGCAATTCAGTTGAG-3′) 和3′端与siRNA 3′端反向互补配对的siRNA特异性序列组成,根据与siRNA配对的寡核苷酸数量 (6或8个) 不同对每个siRNA设计了两种茎环引物(分别为 SLP-N1-6、SLP-N1-8和 SLP-N2-6、 SLP-N2-8)。实时定量PCR上游引物包括用于提高 Tm值并延伸 PCR产物的 5′端通用序列(5′-ACACTCCAGCTGGG-3′) 和3′端siRNA特异序列。MGB探针由5′端6-FAM标记的通用序列 (5′-TTCAGTTGAG-3′) 和 3′端 MGB标记siRNA特异序列组成。选择表达丰度较高且稳定的猪内源性ssc-miR16为内参,监测样本质量、RNA提取及 RT-PCR等过程。所用引物均由南京金斯瑞基因有限公司合成,MGB探针由上海基康基因有限公司合成,标准品ssiN1 (人工合成的siN1,序列见表1) 由TaKaRa公司合成。
1.4 茎环法RT-qPCR
1.4.1 总RNA提取
将获取的抗CSFV的PK-15细胞克隆培养于6孔细胞培养板中,当满度达90%至95%时,弃培养液,用1 mL冷PBS在冰上洗细胞2次,然后按MirVana miRNA isolation kit说明书建议加入 500 μL裂解液裂解后提取总 RNA,以正常PK-15细胞为阴性对照。用NanoDrop 1000测定RNA浓度及质量,分装于−80 ℃保存备用。

表1 siRNA及引物和MGB探针序列Table 1 Sequence of siRNA, primer and MGB probe
1.4.2 茎环法逆转录反应
逆转录反应体系包括50 nmol/L茎环引物,0.25 mmol/L dNTPs,3.33 U/μL逆转录酶,0.25 U/μL RNA酶抑制剂,1倍逆转录缓冲液,10 ng总RNA,用无RNA酶水补至15 μL。反应条件为冰上放置5 min,16 ℃ 30 min ,42 ℃延伸30 min,85 ℃酶灭活5 min,4 ℃保存。所有逆转录反应均为两个重复。
1.4.3 实时定量PCR反应
实时定量PCR反应体系为10 μL PCR预混液,1.5 μmol/L上游引物,0.7 μmol/L通用下游引物,0.2 μmol/L MGB探针,1.2 μL cDNA产物,去离子水补至 20 μL。在 Agilent Stratagene Mx3000P实时定量PCR仪上完成反应,条件为:95 ℃热启动10 min,95 ℃变性15 s,60 ℃退火1 min,在60 ℃收集荧光,共40个循环。所有反应包括阴性细胞对照 (NC) 和空白对照(NTC) 均为2个重复,结果取均值,产物连接T载体测序验证。
1.4.4 标准曲线的绘制及灵敏度分析
为准确定量细胞克隆的siRNA表达水平,以10 mg/L浓度的PK-15细胞总RNA为稀释液10倍梯度稀释已知拷贝数的标准品ssiN1,取8个梯度 (拷贝数从 10到 1.0×108) 进行茎环法RT-qPCR检测绘制标准曲线,计算细胞表达的siRNA拷贝数,并分析此方法检测灵敏度。
1.4.5 特异性分析
因细胞表达数百种与siRNA大小相似的内源性 miRNA,可作为特异性分析材料,所以选择PK-N1、PK-N2和 PK-siC细胞克隆及阴性细胞PK-15和PFF的总RNA为模板进行siN1和siN2的茎环法RT-qPCR检测,以评价检测特异性。
1.5 数据分析
用Agilent公司的Stratagene Mx3000P软件分析 PCR结果。采取手动方式将阈值选定在固定荧光阈值 6000得到各反应管的 Cq值(Quantification cycle),软件自动生成标准曲线。用SPSS软件完成数据统计分析。
2 结果
2.1 抗CSFV PK-15细胞克隆的制备及鉴定
将 G418筛选获得的 PK-15细胞克隆进行neo基因PCR鉴定,均成功扩增出了neo基因片段 (图略),说明细胞克隆均整合了目的基因片段;抗CSFV检测结果如图1所示,IFA结果中阳性细胞克隆荧光比例为5%左右,远远低于对照细胞 PK-siC的 80%~85%,表明有效降低了CSFV抗原的合成;阳性细胞克隆的病毒基因组拷贝数和 TCID50测定结果均显著低于对照细胞(P<0.01),表明有效抑制了病毒基因组复制和成熟病毒粒子装配。以上结果共同表明已成功获得了抗CSFV的PK-N1和PK-N2细胞克隆。
2.2 茎环逆转录引物筛选
应用两种茎环引物分别以相应阳性克隆、PK-15细胞的总 RNA 为模板进行茎环法RT-qPCR检测。为筛选出最佳茎环引物,以具有较高逆转录效率且较低的背景信号为筛选原则,并引入 ΔCq进行量化评价,每种逆转录引物的ΔCq=Cq(阴性细胞对照) –Cq(阳性细胞克隆),ΔCq越大,则逆转录引物具有越高的逆转录效率且产生越低的背景信号。结果如图2所示,siN1检测体系的茎环引物SLP-N1-6的ΔCq(7.09) 远大于SLP-N1-8 (0.32),siN2检测体系的SLP-N2-8的ΔCq(10.48) 远大于SLP-N2-6 (5.93),所以均为最佳的茎环引物。
2.3 特异性分析
特异性分析结果如表 2所示,PK-N1和PK-15的siN2检测结果相同,PK-N2和PK-15的siN1检测结果也相同,说明siN1和siN2两种检测体系不会相互影响,均只能特异性地检测各自目的siRNA;PK-siC和PK-15的siN1检测结果相同,说明乱序的siN1分子不会影响siN1的检测;而两种阴性细胞PK-15和PFF的Cq均在35左右,且远大于阳性克隆 (Cq为25-28),说明检测背景底,可通过 Cq的差异有效地区分阴阳性结果。以上结果共同表明我们建立检测 siN1和siN2的茎环法RT-qPCR受内源性miRNA的干扰很小,具有很高的检测特异性。

图1 抗CSFV PK-15细胞克隆鉴定Fig. 1 Identity results of anti-CSFV cell clones. (A) IFA results of PK-15 cell clones at 72 h after CSFV infection (40×). Mock transfection: stained with CSFV-negative serum. (B,C) Quantity of CSFV genomic RNAs and infectious CSFV production at 48 h and 72 h after CSFV infection. PK-siC: PK-15 cell clones expressing scrambled siN1. ** P<0.01, very signifcant difference with PK-siC control.

图2 茎环引物的筛选结果Fig. 2 Results of screening stem-loop primers. (A) 1, 3: PK-N1 RNA reverse transcribed with SLP-N1-8 (Cq=25.71) and SLP-N1-6 (Cq=28.14); 2, 4: PK-15 with SLP-N1-8 (Cq=26.03) and SLP-N1-6 (Cq=35.23). (B) 5, 6: PK-N2 RNA reverse transcribed with SLP-N2-8 (Cq=25.97) and SLP-N2-6 (Cq=31.82); 7, 8: PK-15 with SLP-N2-8 (Cq=36.45) and SLP-N2-6 (Cq=37.75).
2.4 标准曲线的绘制及灵敏度分析
以ssiN1为标准品绘制标准曲线,结果如图3所示,1~7个梯度有很高的重复性,并能与阴性对照相区分,能检测100个拷贝的siRNA;检测线性范围宽,可达 7个数量级;平行性好(Rsq=0.999),扩增效率高 (Eff.=98.2%);可对目的siRNA进行定量分析。
2.5 PK-N1和PK-N2细胞克隆的siRNA定量检测
以经抗CSFV鉴定的PK-N1和PK-N2细胞克隆各3个进行siRNA定量检测,用2-ΔCq法计算目的 siRNA相对于内参 ssc-miR16的表达水平,如表2所示,结果表明阳性细胞克隆PK-N1和PK-N2均表达了目的siRNA,但每个克隆的siRNA表达水平不同,最高为内参ssc-miR16的 5.54倍,最低的为0.62倍,通过标准曲线计算其拷贝数在 1.0×104左右。产物经测序鉴定正确,表明成功建立了可准确定量检测siN1和siN2的茎环法RT-qPCR。

图3 ssiN1检测动态范围及灵敏度分析Fig. 3 Dynamic range and sensitivity of the ssiN1 assay. (A) Amplification plot of ssiN1 over eight orders of magnitude. 1-8: ssiN1 input ranged from 10 to 1.0×108 copies; NC: negative control; NTC: no template control. (B) Standard curve of the ssiN1.
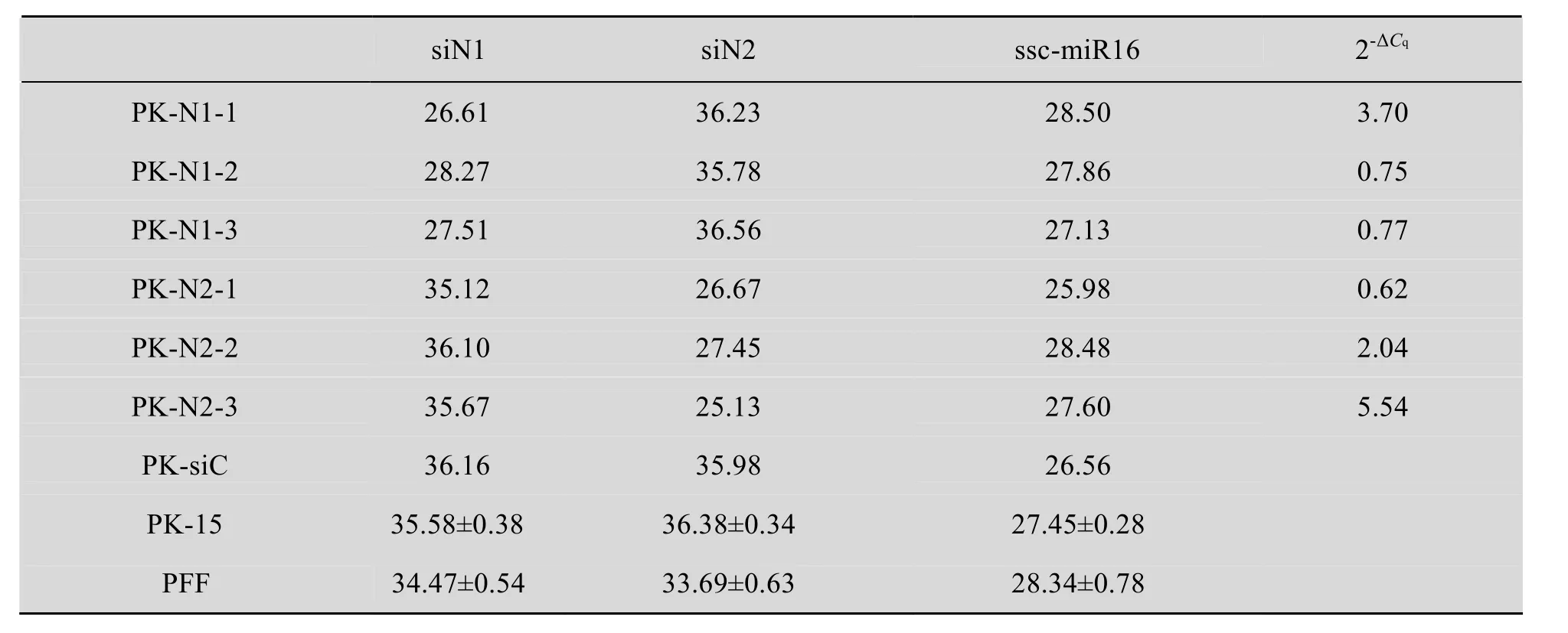
表2 阳性细胞克隆检测及特异性分析结果Table 2 Results of positive cell clones and specificity analysis
3 讨论
目前,RNAi已广泛地应用于抗病毒治疗研究,一般通过测定细胞接毒后病毒基因组拷贝数(Real time RT-PCR) 和蛋白表达水平 (IFA) 及成熟病毒粒子装配水平 (TCID50)[4,6]来评价抗病毒效果,但在某些情况下定量检测siRNA的表达水平是非常必要的。我们曾为确定siRNA四表达载体是否表达了每种siRNA而按ter Brake等[1]的方法构建了siRNA靶基因和报告基因GFP融合表达的报告质粒,与siRNA表达质粒共转染细胞,可通过报告基因的表达来判断siRNA是否表达及其抑制效果[6]。但这种间接的方法应用范围窄,仅能评价siRNA载体的有效性,所以需要建立一种直接检测siRNA的方法,以满足广泛的需要。
目前已有多种可用于检测siRNA的方法,而选择最适的方法是本实验成败的关键。荧光定量PCR是基因表达定量分析的金标准,尽管siRNA的短小对其提出了巨大挑战,但仍有多种实时定量方法被开发出来[9-16],这些方法兼具了实时定量 PCR的高灵敏度、高特异性、准确定量及宽动态范围的实时监测等优点。而茎环法RT-qPCR因创新地设计了茎环引物,其茎部的碱基堆积作用大大增加了RNA-DNA杂交双链的热稳定性,另外,茎环的空间限制作用使之比常规线性引物有更高的特异性,并联合特异性MGB探针,使其成为准确定量siRNA的最佳选择。
在茎环引物的筛选过程中,我们发现对于siN1检测体系 (图2A),用SLP-N1-8逆转录阳性细胞 (25.71) 和阴性细胞对照 (26.03) 的 Cq值基本相同,可能是体系中上游引物与茎环引物产生杂交反应,产生很高的假阳性信号,是不合格的引物;而 SLP-N2-6有较低的背景信号(35.23),可有效区分阴阳性结果,是较理想的逆转录引物。对于 siN2检测体系 (图 2B),无论SLP-N2-6 还是 SLP-N2-8的阴性细胞对照均有较低的背景信号,但 SLP-N2-8的阳性细胞(25.97) 的Cq远小于SLP-N2-6 (31.82),而这种信号不可能来自于较低的背景信号,所以说明SLP-N2-8有更高的逆转录效率,为较佳的逆转录引物。根据以上研究结果我们发现茎环引物的3′端与siRNA配对的碱基个数对实验成败至关重要,配对8个可能会与上游引物重叠导致杂交反应,造成假阳性结果,而配对6个又可能会降低逆转录效率,所以本实验设计两种茎环引物,以实验来筛选效果最佳者,并引入ΔCq来进行量化评价,结果选择了SLP-N1-6和SLP-N2-8为最佳茎环引物。
因为 siRNA和 miRNA均为21 nt左右的RNA分子,所以siRNA的检测一般会受到细胞内源性数百种且表达量较高的miRNA的干扰。我们曾用Shi等[15]的Poly (A) 加尾法RT-qPCR检测目的siRNA,但其阴性细胞对照及水对照均有扩增曲线 (数据未显示),可能是受miRNA的干扰所致,再加上引物二聚体与扩增产物的溶解曲线 Tm大小相近,给阴阳性结果的分析判断带来极大困难。而 Cheng等[17]报道了茎环法RT-qPCR也会产生非特异性曲线,他们设计的107个siRNA检测体系,在以HeLa细胞RNA为阴性对照时,绝大多数体系 (101个) Cq值大于35,有5个Cq值在30至35之间,1个Cq值小于30,而阳性结果均小于 30,所以阴性细胞对照的Cq值大于35可说明体系有较低的非特异性扩增反应,不会影响目的siRNA的检测。为了探明细胞内数百种 miRNA对我们所建立的siN1siN2检测体系的影响,选择了具有独特miRNAs表达谱系的 PK-15和 PFF两种阴性细胞,结果PK-15细胞 (见表2) 的Cq值均大于35,而PFF细胞的Cq值均在33到35之间,说明了细胞的miRNAs表达谱系会对检测有所影响,但这种影响又是微乎其微的 (两种阴性细胞仅相差1到2个Cq,而与阳性细胞相差7到10个Cq),综上结果表明,我们所建立的检测siN1和siN2的茎环法RT-qPCR均有很高的检测特异性。
为了准确定量细胞siRNA的表达水平,曾应用Promega公司的 Riboprobe System-T7系统体外转录的 siN1分子为标准品,但因无法准确测定siN1拷贝数而选择了ssiN1,并用10 mg/L的正常PK-15细胞总RNA为稀释液进行10倍梯度稀释,模拟细胞内的真实环境,充分考虑了细胞内源性miRNA的干扰,以达到准确定量siRNA的目的。
虽然筛选获得的抗CSFV细胞克隆均表达了目的 siRNA,但每个克隆的siRNA表达水平显著不同,这可能是由于外源基因随机地整合到受体细胞染色体的任意位置,产生了位置效应,也可能是随机整合了多个外源基因所致。
本研究建立的茎环法RT-qPCR,可用于准确定量检测细胞抗病毒 siRNA的表达水平,联合IFA等检测病毒水平的方法定量评价RNAi抗病毒的有效性。在抗猪瘟病毒转基因动物的构建过程中,此方法可用于筛选siRNA高效表达的供体细胞克隆,从而提高抗病毒转基因动物的成功率,并可应用于未来转基因动物的抗病毒效果的评价。另外,本方法对siRNA、miRNA等小RNA的检测工作具有很好的参考价值。
REFERENCES
[1] ter Brake O, 't Hooft K, Liu YP, et al. Lentiviral vector design for multiple shRNA expression and durable HIV-1 inhibition. Mol Ther, 2008, 16(3): 557−564.
[2] Wu CJ, Huang HW, Liu CY, et al. Inhibition of SARS-CoV replication by siRNA. Antiviral Res, 2005, 65(1): 45−48.
[3] Ruggli N, Bird BH, Liu LZ, et al. Nproof classical swine fever virus is an antagonist of double-stranded RNA-mediated apoptosis and IFN-α/β induction. Virology, 2005, 340(2): 265−276.
[4] Xu XR, Guo HC, Xiao C, et al. In vitro inhibition of classical swine fever virus replication by siRNAs targeting Nproand NS5B genes. Antiviral Res, 2008, 78(3): 188−193.
[5] Li JN, Guo HC, Shi ZX, et al. In vitro inhibition of CSFV replication by retroviral vector-mediated RNA interference. J Virol Methods, 2010, 169(2): 316−321.
[6] Li JN, Dai YJ, Liu S, et al. In vitro inhibition of CSFV replication by multiple siRNA expression. Antiviral Res, 2011, 91(2):209−216.
[7] Pall GS, Codony-Servat C, Byrne J, et al. Carbodiimide-mediated cross-linking of RNA to nylon membranes improves the detection of siRNA, miRNA and piRNA by Northern blot. Nucleic Acids Res, 2007, 35(8): e60.
[8] Várallyay É, Burgyán J, Havelda Z. Detection of microRNAs by Northern blot analyses using LNA probes. Methods, 2007, 43(2): 140−145.
[9] Jiang M, Arzumanov AA, Gait MJ, et al. A bi-functional siRNA construct induces RNA interference and also primes PCR amplification for its own quantification. Nucleic Acids Res, 2005, 33(18): e151.
[10] Raymond CK, Roberts BS, Garrett-Engele P, et al. Simple, quantitative primer-extension PCR assay for direct monitoring of microRNAs and short-interfering RNAs. RNA, 2005, 11(11): 1737−1744.
[11] Ro S, Park C, Jin JL, et al. A PCR-based method for detection and quantification of small RNAs. Biochem Biophys Res Commun, 2006, 351(3): 756−763.
[12] Varkonyi-Gasic E, Wu R, Wood M, et al. Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods, 2007, 3: 12.
[13] Sharbati-Tehrani S, Kutz-Lohroff B, Bergbauer R, et al. miR-Q: a novel quantitative RT-PCR approach for the expression profiling of small RNA molecules such as miRNAs in a complex sample. BMC Mol Biol, 2008, 9(1): 34.
[14] Stratford S, Stec S, Jadhav V, et al. Examination of real-time polymerase chain reaction methods for the detection and quantification of modified siRNA. Anal Biochem, 2008, 379(1): 96−104.
[15] Shi R, Chiang VL. Facile means for quantifying microRNA expression by real-time PCR. Biotechniques, 2005, 39(4): 519−525.
[16] Chen C, Ridzon DA, Broomer AJ, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res, 2005, 33(20): e179.
[17] Cheng AG, Li M, Liang Y, et al. Stem-loop RT-PCR quantification of siRNAs in vitro and in vivo. Oligonucleotides, 2009, 19(2): 203−208.
[18] Yu XH, Tu CC, Lu HW, et al. DNA-mediated protection against classical swine fever virus. Vaccine, 2001, 19(11/12):1520−1525.
[19] Tang FC, Hajkova P, Barton SC, et al. MicroRNA expression profiling of single whole embryonic stem cells. Nucleic Acids Res, 2006, 34(2): e9.
