基于结合位点的辅酶A结合蛋白家族的分类
2011-11-30刘振明金宏威张亮仁
樊 迪 刘振明, 金宏威 张亮仁
(北京大学药学院,天然药物及仿生药物国家重点实验室,北京100191)
基于结合位点的辅酶A结合蛋白家族的分类
樊 迪§刘振明§,*金宏威 张亮仁*
(北京大学药学院,天然药物及仿生药物国家重点实验室,北京100191)
发展了一种基于分子相互识别的蛋白质分类方法,应用数据挖掘策略与统计学聚类,根据辅酶A (coenzyme-A,CoA)结合蛋白的结合模式特征数据,通过对比和分析多种分类方法对该体系的分类准确度,对这类体内重要的蛋白进行了分类方法学研究,选择了最优的两步聚类法.本研究工作设计和建立了一个分类参数,可以简洁有效地评价出各个结合特征的显著性与重要性,并以此为依据从所有特征中筛选出决定性的特征变量.研究结果所得到的CoA结合蛋白的三个分类,都具有显著的氢键与疏水结合特征;CoA可以与多个生物活性关键氨基酸残基形成氢键作用.这些相互作用的共性及分类上的差异,说明了配体与不同受体相互作用过程中结合模式上的细微差别,对于以CoA结合蛋白为靶点的选择性调控分子设计具有重要的参考意义与指导作用.
辅酶A;蛋白质分类;结合模式;聚类分析;泛酰巯基乙胺链
1 引言
蛋白质与配体的结合模式特征及分类研究对于阐述体内生理过程及药物发现与选择性改造具有重要的指导意义.作为化学基因组学的一种重要研究手段,从配体和受体的相互作用出发,研究蛋白质的功能分类,不仅可以揭示出分类特征与生物学功能之间的关系,同时也可以进一步明确结合位点的特征,为基于该类靶点的药物设计提供指导.1-3目前,基于结合位点的蛋白质分类功能学研究已经有了一些成功的研究工作和进展.4-13
蛋白质的分类算法可以分为有监督的分类和无监督的聚类两种.有监督分类的代表为机器学习,其中以支持向量机(SVM)应用最为广泛;14-18无监督聚类的代表为Kmeans、系统聚类等.19-21不同的研究小组对各种聚类算法进行了深入的比较研究. Markowetz等18对268个蛋白基于序列进行分类,使用SVM方法得到结果的准确率明显高于其他六种方法,尤其是在高维的情况下,有效地减少了错误率.Kertész-Farkas等22评测了多种方法的交叉验证,包括SVM、神经网络、随机森林等,验证得出在多种分类体系中SVM有着非常突出的表现.

图1 辅酶A(CoA)的化学结构Fig.1 Chemical structure of coenzyme-A(CoA)moleculeThe structure contains three fragments:adenosine,ribose,and 4-phospho pantetheine arm.
CoA是生物体内参与乙酰化反应的重要辅酶,在糖、蛋白质和脂肪的代谢过程中具有重要的作用. CoA的结构如图1所示,从左至右可以分为三个部分:4′-磷酸泛酰巯基乙胺链、糖环和腺苷.截止到2010年4月底,Protein Bank Database(PDB)数据库中已经发表的CoA结合蛋白晶体结构共有218个,在功能上涵盖了很多重要的生理和病理过程.23从PDB已发表的晶体结构上看,CoA在与不同蛋白受体结合时,可以采取不同的碱基或者糖环取向.特别是4′-磷酸泛酰巯基乙胺链构象的多样性,充分说明了同一配体在与不同靶蛋白结合时可以采取多个低能空间范围内的药效构象.对这种现象和规律的研究有助于我们理解配体-受体结合过程中自由能焓变与药效构象选择之间的关系.此外,CoA分子与其他一些核苷类分子如烟酰胺腺嘌呤二核苷酸磷酸(NADP)、黄素腺嘌呤二核苷酸(FAD)以及三磷酸腺苷(ATP)类似,都含有一个二磷酸腺苷(ADP)的结构单元.这类结构单元尽管在多类核苷分子中出现,但是却很少作为反应单元直接参与到生化反应中来,更多的时候是作为一种协助的角色出现.24,25NADP与FAD作为辅因子在与蛋白进行结合时通常采取类似的结合取向模板与蛋白中的经典Rossman折叠区域结合.但是这种空间取向的平面与CoA分子是大相径庭的.对CoA及其结合蛋白进行研究,将有助于阐述其结构和功能之间的内在联系.
在已有研究工作的基础上,我们建立和发展了一种新的蛋白质分类流程与方法,通过深度挖掘提取CoA与蛋白的结合位点信息得到相互作用数据,利用两步聚类的方法对CoA结合蛋白家族进行了分类研究.聚类结果及每一个类别的特征有助于对CoA及其结合蛋白的相互关系做更进一步的了解,为相关的分子设计提供指导与帮助.
2 研究方法
本研究工作分为四个部分:(1)收集和处理数据样本,对从PDB数据库中获取的晶体复合物进行处理,分离得到受体和配体的结构坐标文件,并转化为聚类计算所需要的文件格式;(2)对处理好的晶体文件进行数学描述,将受体和配体之间相互作用的有、无和程度用量化的方式表示出来;(3)采用已知的数学和统计学方法,分析量化好的数据,对以上述数据为特征变量的蛋白质进行分类,并且进行验证,选择最优方法与最优解;(4)对分类的结果进行分析,包括类别分析,特征变量分析,与其他分类体系对照等.具体流程如图2所示.
2.1 数据来源
从PDB蛋白质结构数据库中以CoA为检索词搜索数据库,得到259个CoA结合蛋白的晶体文件;以含有完整CoA结构、不重复、分辨率高优先为原则,筛选出66个非冗余CoA结合蛋白.使用Sybyl 6.91程序26和Discovery Studio 2.0软件包27提取复合物结合口袋信息,然后采用Pocket程序28进行数据挖掘,得到CoA与20种天然氨基酸的氢键和疏水相互作用共72项特征数据.其中氨基酸特征60个,分别为与20种氨基酸形成氢键,CoA作氢键给体、受体或两者兼有,分别用A、D、A/D表示;形成疏水作用的有12种氨基酸.详细数据请参见本研究论文的补充材料部分(Supplementary materials: available free of charge via the internet at http://www. whxb.pku.edu.cn)

图2 对CoA结合蛋白进行分类研究的实验设计流程图Fig.2 Designed flow chart for CoAbinding proteins classification
2.2 聚类方法比较选择
聚类分析指的是将集合中的对象按照相似性分为多类.聚类方法可以不指定分类数目,无需学习;算法根据特征变量,自动寻找相似性较高的元素,并将之作为同一类.研究工作中主要应用和比较了K-means法、两步聚类、系统聚类和SVM四种聚类方法.
K-means法可译为K均值法,又名快速聚类,是一种经典的聚类方法,具体过程是:选择聚类数k个值作为初始聚类中心,由n个待聚类变量组成n维空间,按照每个点距聚类中心最小原则,将各点划入中心周围,完成第一次迭代,接下来根据迭代计算平均值,将每一类的均值(共k个)放入n维空间中,再作为新的聚类中心进行第二次迭代,如此循环,直至达到指定迭代次数或中止迭代条件.K-means法的缺陷在于选择聚类中心的随机性,以及异常值对聚类结果的影响.
两步聚类法的优势在于既可以处理连续变量,又可以处理分离变量,能自动确定最佳聚类数目,对大数据集的处理速度快.首先是逐个扫描样本,并计算每个样本与已扫描样本的距离并归类,归为已有类或生成新的类.然后,依据第一步的分类结果,根据各分类之间的距离,对各个类别进行合并,并按照指定标准停止合并.最佳聚类数目的确定,需要两个步骤.首先是使用贝叶斯信息量准则(BIC)或赤池信息量准则(AIC)初步估算聚类数目,然后,根据初步估算的结果,测算聚类之间的最近距离,并进行修正.两步聚类从一定程度上弥补了K-means的缺陷.
系统聚类又名分层聚类,主要适用于样本量不是很多的聚类分析,属于比较泛用的聚类方法,有两种方向相反的聚类过程.分解法是先把全部样本看作一个大类,然后根据距离和相似性逐层分解为小类;凝聚法是先把每个样本视为一类,根据距离和相似性逐渐合并.系统聚类法提供了多种聚类算法和量度的组合可供选择,本研究工作选取了准确率最高的三种组合.
SVM方法的基本思想是:寻找一个超平面H(d),该超平面可以将训练集中的数据分开,且与类域边界的沿垂直于该超平面方向的距离最大,故SVM法也被称为最大边缘(maximum margin)算法.其中,起主导作用的是“支持向量”,非支持向量的量.近年来,很多方法学对比的研究工作表明,一般情况下,在机器学习分类算法中,SVM具有最高的准确率.本研究中,我们采用由台湾大学林智仁编写的Libsvm 2.9模式识别和回归软件包29来进行SVM的分类.SVM方法作为有监督分类的代表,在实际研究中表现出了较高的准确率,在本研究中作为聚类分析方法的参照.
2.3 特征的筛选
在本研究体系中,作为分类依据的特征共有72个,但实际上起主导作用的并非全部.将这些特征筛选出来,就是把结合位点特征挑选出来的过程.
在统计学上,可以使用相关性分析、卡方检验等方法来对各个变量的显著性和相关性进行研究.对于该体系,我们发展了一种简易的方法,可以有效地对分类特征进行筛选.该方法对一个体系中两个分类的分类特征的显著性有比较好的区分效果.
对于分为甲、乙两组的每一个特征变量:

其中:F为该方法的系数,数值区间(-∞,+∞);a为该特征变量在甲组中出现的频度;b为该特征变量在乙组中出现的频度;X为该特征变量在甲组中的绝对数量;Y为该特征变量在乙组中的绝对数量.a/b,即为该特征变量在两个分组中出现频率之比,比值大于1表示在甲组中的出现率高于乙组,可判定为甲组特征;比值小于1则属于乙组特征.取自然对数ln,可以将甲乙组的特征归属用正负来表示.由于采用比值,如果计算出a和b的绝对数量过小,会导致假阳性的出现,因此加入修正|X2-Y2|,即绝对数量之平方差的绝对值.平方差可因式分解为(X+Y)(X-Y),和可以增大绝对数量的权重,使两个分组出现频率皆很低但比值却很大造成假阳性的特征;差可以削减绝对数量较多,但两组数量接近造成假阳性的特征;取绝对值目的是保证取自然对数的结果的正负不改变,从而对正确的筛选起到积极作用.这样,就可以基本完全排除假阳性的出现.
只有一组独有的特性直接作为特性处理,不参加运算,然后将共有特性排除,不同分组之间互相计算系数.正值越大,表示该特征变量越倾向于甲组特性;负值越大,表示该特征变量越接近乙组特性.通过对照数值与具体结合特征,判定F绝对值在30以上的特征变量是在分类中起主导作用的特征变量.
3 结果与讨论
3.1 CoA结合蛋白的功能分类
与核糖核酸类似物(包括COA、ATP、ADP等)相结合的蛋白质,可以从配体与蛋白质作用的位点和功能分为两类:(1)催化作用,(2)合成其他衍生物时作为底物或产物存在.以CoA为例,脂肪酸响应性转录因子(PDB编号:1H9G)与CoA的复合物中, CoA结合在催化位点上,发挥催化作用;而在HMG-COA还原酶(PDB编号:1DQA)复合物中, CoA是以产物的形式存在的.以此为依据,将66个蛋白分为两类,其中CoA起催化作用的蛋白为44个(A组),作为产物或底物的22个(B组).本文后继的方法学研究和聚类分析都是建立在这套数据的基础之上的.
3.2 聚类分析的结果与聚类方法比较
分类完成之后,下一步是确认该聚类方法的可信度,即聚类特征是否在体系内能够自洽;如果自洽,则证明该结果可信.我们采取两种方式使用SVM方法与其他聚类分析方法作比较.其一,随机选择51组数据作训练集,测试剩余15组;其二,进行交叉验证,将数据随机分为多组,分别互相训练与测试,取得平均的准确率.SVM的五重交叉验证结果,印证了国内外很多工作的结论,即SVM的高准确性.但人为将数据分为测试集和训练集后,其准确率便会大打折扣,明显低于期望值.具体结果如表1所示.
分析原因,从交叉验证的原理看,交叉验证是多次训练与测试得到的平均结果;另一方面,随机挑选训练集合测试集并预测.与以往成功的实例相比,本实验66组数据的样本量相对较小.前者为多次学习过程的平均,后者仅有一次,故准确率差距很大.然而,SVM有监督分类的本质,决定了其在本次实验中只能作为参照出现.SVM交叉验证的结果,表明了在CoA结合蛋白中,CoA所处的地位不同,其结合位点的结合特征也有着显著的特异性.
K均值法,对两个分类的预测准确率均在60%以下.本研究工作所选取的数据体系比较复杂,维度为72,而由于K均值法的缺陷,即随机性和异常值的影响,在高维度中,聚类中心的偏差尤其明显,导致难以预测出正确的结果.
系统聚类法提供了多种算法和量度的组合.经过逐一实验对比,我们选出了表1中的三个组合.组内联结算法和余弦量度的组合,对催化组有较高的辨识度,但对底物组的结果却是所有准确率数值中最低的.另外两种组合,总体准确率低于K均值法.
由表1可知,两步聚类算法在无监督分类方法中的准确度是最高的.另外,两步聚类法除克服了K均值法的缺陷外,同时还具有可以选择让其自动确定分类数目的特点,为聚类分析工作带来一定的便利.因此,我们尝试用不指定分类数目的两步聚类法.CoA结合蛋白的最终聚类树如图3所示.可以看到,66个蛋白被自动聚成了两类,数目分别为21和45个.其中,含21个蛋白的分类,包含19个催化型和2个底物型.由此说明催化组和底物组在结合位点上具有某些显著的差异.被分到含45个蛋白聚类中的催化组蛋白,与被单独分类出来的催化型CoA结合蛋白有一定程度的差异,在某些特征上与底物组近似.将催化组单独提出,使用不指定聚类数目的两步聚类法,进一步分别得到数目为26和18的两个亚类.最终,66个蛋白可以被分为三组:催化组1 (A1)、催化组2(A2)和底物组(B),数目分别是26、18和22个.

表1 几种聚类方法准确率的比较Table 1 Accuracy of several cluster analysis methods used in this research work
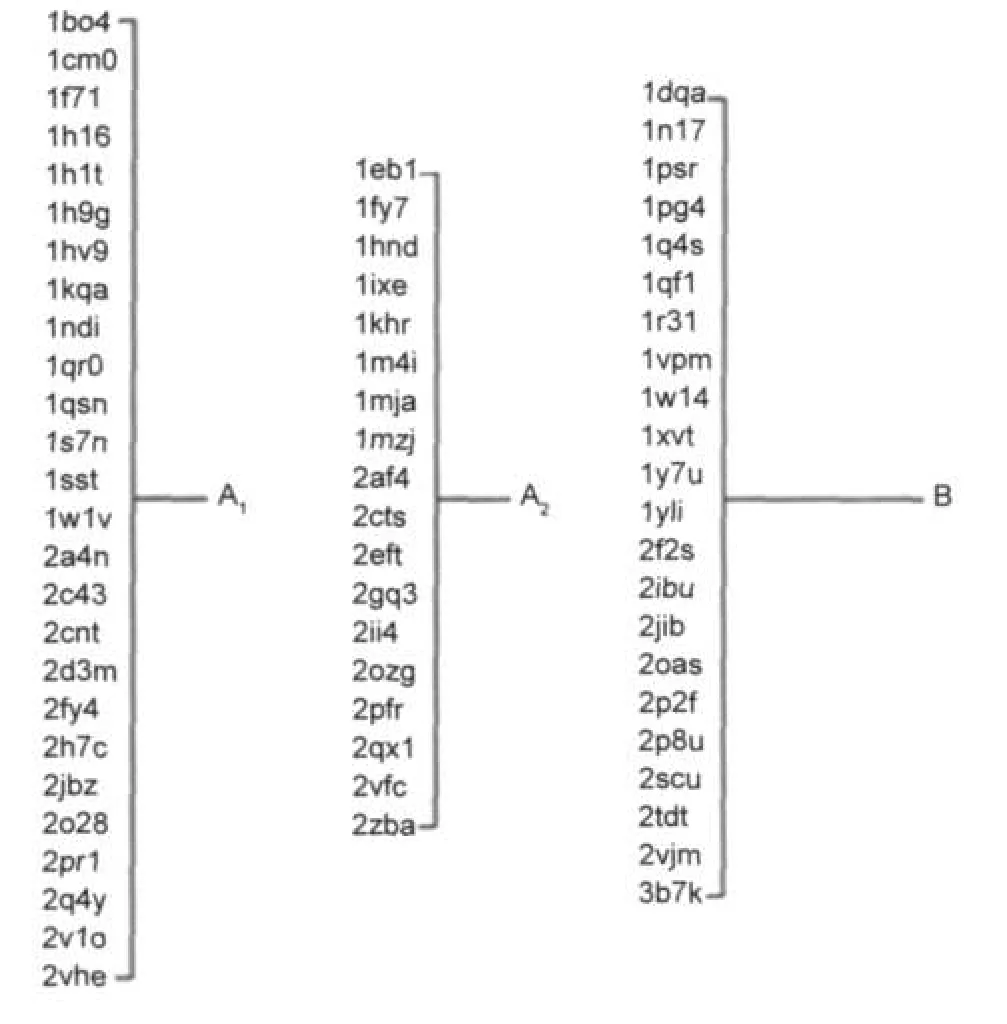
图3 基于结合位点特征的CoA结合蛋白的聚类结果Fig.3 Classification result of CoAbinding proteinsbased on substrate binding patterns
3.3 结合位点特征分析
66个蛋白中,有48个与ARG有氢键作用,39个与LYS有氢键作用,氨基酸残基作氢键给体.CoA与ILE、LEU和PHE形成疏水作用的蛋白也占多数,分别是39、44、37个.这些特性,由于绝对数量过大,用本方法计算,将属于假阳性结果.即F值的绝对值很大而实际并不能归类为特征.由于它们是作为CoA结合蛋白的共性特征存在的,可以为鉴别工作带来帮助.例如,鉴别一个蛋白质是否可以与CoA及其类似物结合,可以观察结合口袋中是否含有这些氨基酸残基.催化组与底物组,以及催化组内部两个亚类的结合特征打分结果如表2所示.
表中数值的绝对值越高,表明特性越明显.正值表示该项为催化组的特征,负值表示该项为底物组的特征.由表可见,催化组与ASP、GLY、TYR发生氢键作用的较多,与LYS发生疏水作用的较多;底物组较多与HIS产生氢键和疏水作用.由表2可知,催化组与ASP、GLY、TYR发生氢键作用的较多,与LYS发生疏水作用的较多;底物组较多与HIS产生氢键和疏水作用.
催化组中,A1组与HIS、SER、THR发生氢键作用的较多,与LYS发生疏水作用的较多;A2组与ASN、SER发生氢键作用的较多,与MET发生疏水作用的较多;二者均与SER有氢键作用,不同的是, A1组以氢键给体出现,A2组既可作给体也可作受体.
3.4 与其他分类体系之关系
我们进一步比较和研究了所得到的66个CoA结合蛋白的分类结果与按照已有的折叠模式(SCOP分类)30,31分类以及酶催化的化学反应类型(EC编号)分类32的异同,如图4所示(图4(A)是66个蛋白中部分按照SCOP的四大折叠类分类的结果;图4(B)是按照EC编号进行分类的结果,在本体系中,催化组大部分蛋白都具有EC编号,但底物组只有一个蛋白具有EC编号,因此这里主要对催化组进行分析).
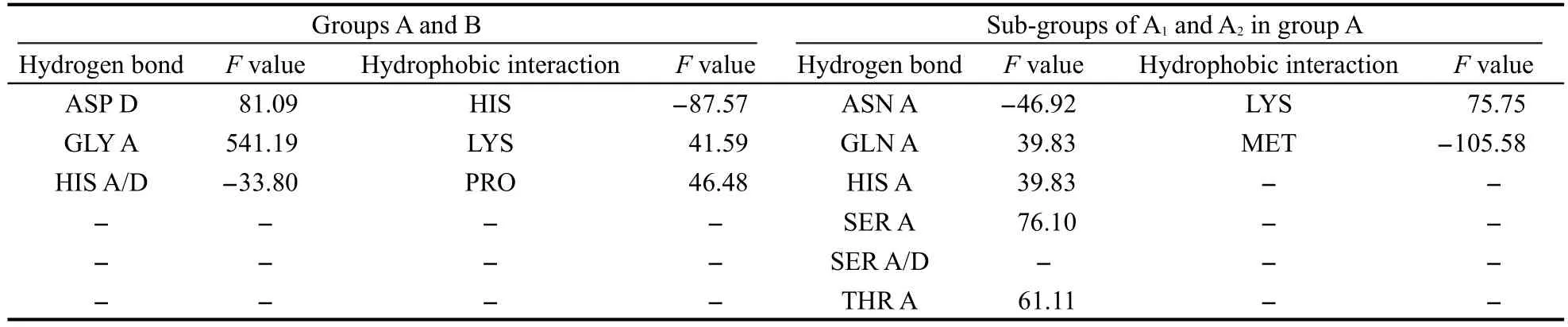
表2 聚类结果的结合位点特征分析Table 2 Binding patterns analysis with the clustering results
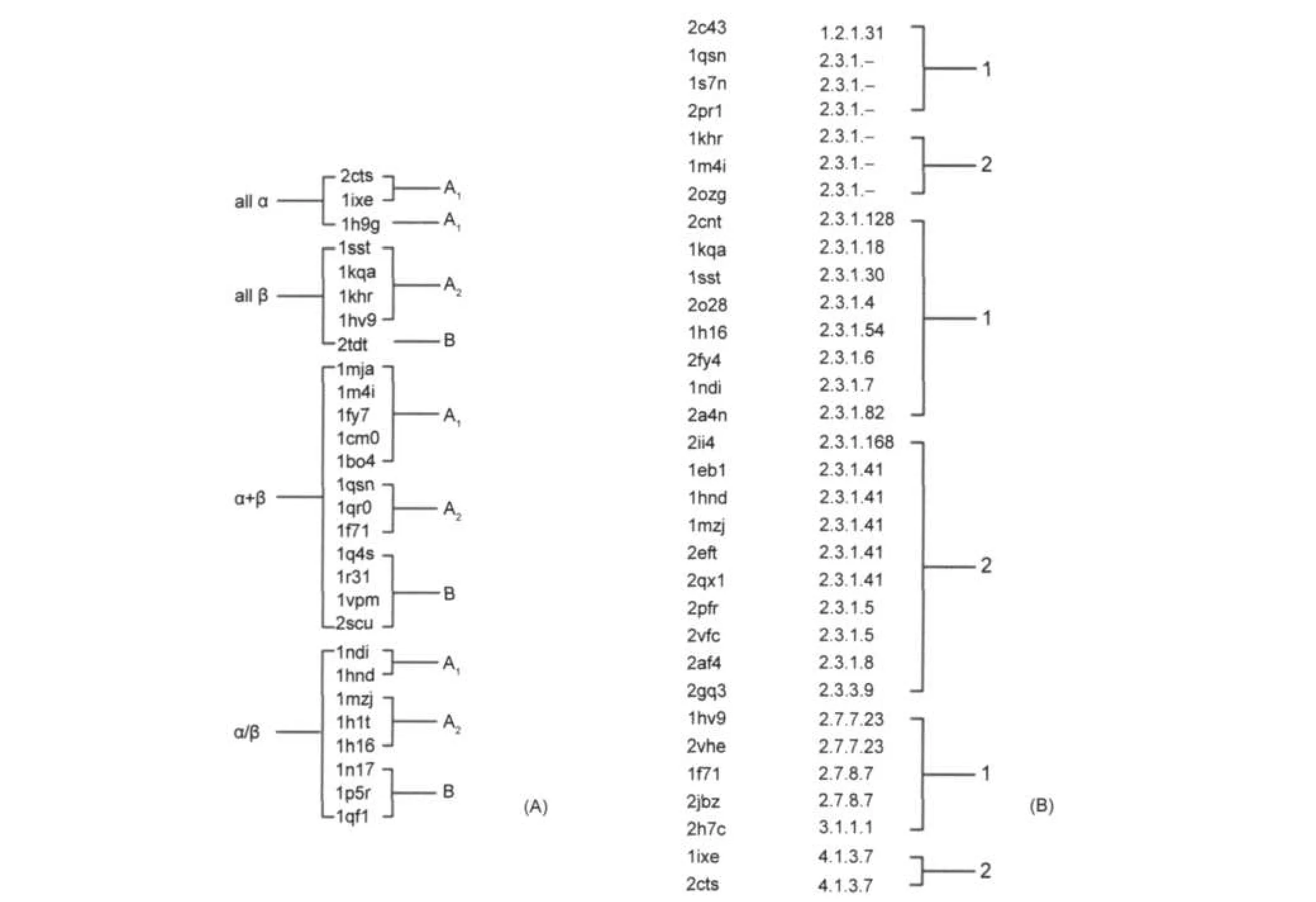
图4 CoA结合蛋白的折叠模式(A)及酶催化的化学反应类型(B)聚类结果Fig.4 Clustering results based on fold of CoAbinding proteins(A)and types of chemical reactions catalyzed by enzyme(B)
由图4(A)可以看出,A组(催化类)包含了all α、all β、α/β、α+β四个折叠类,B组(底物类)不包含all α类型,同时只含有一个all β类蛋白质.而在A1和A2组中,A1组不含all β类.因此可以得出结论,本分类方法与基于蛋白整体序列和结构的分类是迥异的.
EC编号全称为Enzyme Commission number,是依据酶催化的化学反应类型的一种蛋白质分类方法.因此可以说,EC是基于功能的蛋白质分类方法. EC的结构如2.3.1.5,从左至右分别是四个分类层次.第一位是最上级的层次,包括氧化还原酶、转移酶、水解酶、裂合酶、异构酶、合成酶,编号分别是1-6.在本体系中,催化型结合蛋白大部分具有EC编号,底物型结合蛋白只有一个蛋白具有EC编号,故主要对催化型结合蛋白进行分析.从图4(B)中可以看出,编号相同的蛋白质都被正确地分到了同一组.绝大多数催化组蛋白质属于2类,即转移酶,而其中的大部分属于2.3.1类,该类是催化氨酰基以外反应的酰基转移酶.在这类蛋白中,第四位编号有: 4、5、6、7、8、18、30、41、50、82、128、168几种,其中5、8、41和168被划为催化组A2类,其余为催化组A1类.由此可推论,基于结合位点特征的CoA结合蛋白的分类归属,与蛋白质的功能关系非常密切.
使用SVM机器学习方法对SCOP的分类结果进行验证.首先,将28个在SCOP中按照折叠子分类的单提取出来,将all α标记为1类,all β标记为2类,α/β标记为3类,α+β标记为4类.然后使用Libsvm进行五重交叉验证,即将所有数据随机分为5类,互相训练和测试,重复多次,最终计算出平均的准确率.最终,准确率是64.29%.这个数字表明,体系中的分类变量无法支撑SCOP的分类结果.说明基于结合模式的分类方式完全不同于基于蛋白的分类.进一步证明结合模式与结合位点特征,与蛋白质的结构关系并不紧密,结合位点特征相似的蛋白质,其外部结构差异可能会非常大.
3.5 构象研究

图5 以腺苷为模板对CoA分子进行叠合的结果Fig.5 Structure aliment of CoAbases on adenosine fragment
我们尝试从组成CoA分子的三个部分出发,使用Sybyl 6.91程序,对66个复合物中的CoA分子的构象进行叠合.我们首先选择腺苷碱基部分作为叠合模板,因为这是CoA分子中最为刚性的结构部分.但是叠合的结果发现,当腺苷环固定后,分子其他部分的取向变得非常的无序(如图5所示),这从某种程度上说明在CoA分子与蛋白质结合时,腺苷环可能不构成主要的铆定结构.
随后,我们以糖环为模板,将三个亚类蛋白中的CoA构象进行叠合,如图6所示.可以看到,从整体来看,以糖为模板,糖环构象基本无出入,但糖环上的磷酸基构象很多样,长链的分布无规律性.
值得注意的是嘌呤环的取向.由图6可以看到,在糖环重合的情况下,全部CoA构象中的嘌呤环的取向基本一致,在糖环平面之上,绝大多数嘌呤环为同一方向,有极少数取向相反.底物组蛋白全部遵循这个规律,为同一取向,但催化型中,有少数例外.例外的蛋白为:A1组的1CM0的其中一个亚基和1S7N,A2组的1H9G和2H7C.
1CM0,p300/CBP相关因子,重要的组蛋白乙酰转移酶.图7中显示的是取向特殊的亚基A的结合情况,结合口袋为开放式,CoA蜷曲状填充入结合口袋,可以看到结合口袋附近并未填充满,可以作为潜在的分子设计靶点.嘌呤环取向不同于其他,是与图中所示水分子形成水桥所致,溶剂效应在构象的形成中起了一定的作用.另一个亚基B的嘌呤环周围并无水分子,取向是与总体规律相同,因为就分子斥力而言,嘌呤环的这种取向更加稳定.
PDB编号为2H7C的结构是人的羧酸酯酶,是一个含有6个相同亚基的多聚体(如图8所示).结合口袋较为狭窄,长链完全伸入结合口袋的深部.同样,由于水分子的作用,嘌呤环与蛋白质桥接,导致嘌呤环构象扭转.由此可得出结论,这几个特例是溶剂造成的.这也提示我们在分子设计中,应当充分考虑溶剂对结合的影响.

图6 以糖环为平面对CoA构象进行叠合Fig.6 Superimposition of CoAbased on the ribose ring(A)superimposition of CoAconformations binding with catalytic site;(B)superimposition of CoAconformations binding with substrate site; (C)A1sub-class of CoAconformations binding with catalytic site;(D)A2sub-class of CoAconformations binding with catalytic site

图7 1CM0的A亚基结合口袋(a)与B亚基结合口袋(b)Fig.7 Binding sites of CoAwithAsubunit(a)and B subunit(b)from the crystal structure 1CM0
在CoA的结构中,长链部分是多样性最强的. CoA可以与多种具有不同结构与功能的蛋白质分子结合,由于这些蛋白质分子的结构和功能截然不同,结合位点也具有多样性,因此CoA与之结合时,通过采用多种不同构象与之适应,从而发挥不同的调控作用.CoA的长链属于柔性结构,因此在构象的变化中起到了主导作用.由CoA的这种现象可以看出,配体可以通过改变自身的构象,来起到调控不同结构和功能的蛋白质的作用.

图8 人羧酸酯酶(PDB:2H7C)中CoA的结合位点Fig.8 Binding sites of CoAwith human carboxylesterase (PDB entry:2H7C)

图9 根据泛酰巯基乙胺链为模板对CoA分子进行叠合后得到的两种情况Fig.9 Superimposition of CoAbased on pantetheine arm with N24,N28 and C20 fixed(A)CoAbinding with N-acetyltransferase folding protein family; (B)CoAbinding with proteins belongs to single-stranded, left-handed and beta-helix fold family
事实上,CoA分子识别和转运酰基基团的主要功能是通过泛酰巯基乙胺链上的巯基完成的.在生化反应中,CoA分子首先通过巯基基团结合一个酰基.由于碳硫键具有很高的能量,因此很容易后继将酰基基团转移给相应的受体.在泛酰巯基乙胺链上,多个原子参与了CoA分子和蛋白受体之间的相互作用,在其中起到氢键给体或者受体的作用.
我们重新按照泛酰巯基乙胺链为模板进行了叠合,结果发现CoA分子的空间取向可以按照长链的构象分为几类,处于同一分类中的蛋白有可能具有类似的动力学转运机制.在转运的过程中,泛酰巯基乙胺链会转动到一定的取向上,然后结合到蛋白质高度保守的活性位点区域.图9是将侧链上N-4P, N-8P和C-12P作为叠合中心进行分子叠合后得到的两种情况,其中图9(A)中CoA分子对应的蛋白全部来自于N-乙酰转移酶折叠蛋白家族,图9(B)中的蛋白则全部属于左手β螺线管折叠类型,这在某种意义上与SCOP的分类又有所类似.
4 结论
利用数据挖掘策略和统计学分析方法探索出了一条从数据收集、挖掘到分析的策略并进行了针对CoA结合蛋白的分类研究工作,为基于结合位点的蛋白质分类工作的具体操作过程和结果分析提供了一条有效途径.
本研究验证了在对CoA结合位点特征数据的分析中,两步聚类法是一种简单而准确的聚类分析方法,可以高准确率地将体系进行聚类,在类似的蛋白质分类工作中具有很高的实用性.基于结合模式的CoA结合蛋白分类,在分类过程和结果上,都不同于传统的基于序列的分类体系,而且相差甚远.但是这种分类体系和基于功能的分类有着微妙的联系.
在分类结果的分析上,建立了一个新创的系数,能够比较方便而有效地评价两分类中各个分类特征的重要程度,并对寻找分类之间的共性和特性具有较大的帮助,具有较强的实用价值.CoA结合蛋白在结合上具有明显的共性和特性,其中包括CoA分子中磷酸基结构与结合口袋中碱性氨基酸的氢键作用,长链上的羰基与氨基酸残基中的氨基相互作用等,可以在药物设计时,充分考虑到这些特性,并将之应用到实际工作中.同时,CoA在结合位点,在构象上也有一定的规律,嘌呤环具有趋向性特例显示,溶剂因素对配体构象有较大影响,设计分子时应当充分考虑这方面的影响.
(1)Andersson,C.D.;Chen,B.Y.;Linusson,A.Proteins 2010,78, 1408.
(2) Gold,N.D.;Jackson,R.M.J.Chem.Inf.Model.2006,46,736.
(3)Arnold,J.R.;Burdick,K.W.;Pegg,S.C.H.J.Chem.Inf. Comp.Sci.2004,44,2190.
(4) Hoppe,C.;Steinbeck,C.;Wohfahrt,G.J.Mol.Graph.Model. 2006,24,328.
(5) Gold,N.D.;Jackson,R.M.J.Mol.Biol.2006,355,1112.
(6) Izrailev,S.;Farnum,M.A.Proteins 2004,57,711.
(7) Liu,Z.M.;Li,B.;Lai,L.H.Acta Phys.-Chim.Sin.2005,21, 1143.[刘振明,李 博,来鲁华.物理化学学报,2005,21, 1143.]
(8) Cappello,V.;Tramontano,A.;Koch,U.Proteins 2002,47,106.
(9) Kinnings,S.L.;Jackson,R.M.J.Chem.Inf.Model.2009,49, 318.
(10) Doppelt-Azeroual,O.;Delfaud,F.;Moriaud,F.Protein Sci. 2010,19,847.
(11) Li,B.;Liu,Z.M.;Zhang,L.G.;Lai,L.H.J.Chem.Inf.Model. 2009,49,1725.
(12) Balakin,K.V.;Tkachenko,S.E.;Lang,S.A.J.Chem.Inf. Comp.Sci.2002,43,1332.
(13) Patel,R.Y.;Doerksen,R.J.J.Proteome Res.2010,9,4433.
(14) Cai,C.Z.;Han,L.Y.;Ji,Z.L.Nucl.Acids Res.2003,31,3692.
(15) Cai,C.Z.;Wang,W.L.;Sun,L.Z.Math.Biosci.2003,185,111.
(16)Shamim,M.T.A.;Anwaruddin,M.;Nagarajaram,H.A. Bioinformatics 2007,23,3320.
(17) Vapnik,V.N.The Nature of Statistical Learning Theory,1st ed.; Springer-Verlag:New York,1999;pp 30-39.
(18) Markowetz,F.;Edler,L.;Vingron,M.Biometrical J.2003,45, 377.
(19) Shen,H.B.;Yang,J.;Liu,X.J.Biophys.Res.Commun.2005, 334,577.
(20) Kong,J.H.;Fish,D.R.;Rockhill,R.L.J.Comp.Neurol.2005, 489,293.
(21) Liu,Y.;Li,X.Q.;Xu,H.S.;Qiao,H.Acta Phys.-Chim.Sin. 2009,25,2558.[刘 岳,李晓琴,徐海松,乔 辉.物理化学学报,2009,25,2558.]
(22) Kertész-Farkas,A.;Dhir,S.;Sonego,P.;Pacurar,M.;Netoteia, S.;Nijveen,H.;Kuzniar,A.;Leunissen,J.A.M.;Kocsor,A.; Pongor,S.J.Biochem.Biophys.Methods 2008,70,1215.
(23)Welcome to Brookhaven Protein Data Bank.http://www.rcsb. org(accessed,2010).
(24) Leonardi,R.;Zhang,Y.M.;Rock,C.O.Prog.Lipid Res.2005, 44,125.
(25) Rudel,L.L.;Lee,R.G.;Cockman,T.Curr.Opin.Lipidol.2001, 12,121.
(26) Sybyl 6.91.http://www.tripos.com.Tripos;USA,2001.
(27) Discovery Studio 2.0.http://www.accelrys.com/.Accelrys; USA,2008.
(28) Chen,J.;Lai,L.H.J.Chem.Inf.Model.2006,46,2684.
(29) Chang,C.;Lin,C.LIBSVM,Version 2.3;Software available at http://www.csie.ntu.edu.tw/~cjlin/libsvm.2001.
(30) Murzin,A.G.;Brenner,S.E.;Hubbard,T.;Chothia,C.J.Mol. Biol.1995,247,536.
(31)Andreeva,A.;Howorth,D.;Chandonia,J.M.;Brenner,S.E.; Hubbard,T.J.P.;Chothia,C.;Murzin,A.G.Nucleic Acids Res. 2008,36,D419.
(32) Gasteiger,E.;Gattiker,A.;Hoogland,C.;Ivanyi,I.;Appel,R. D.;Bairoch,A.Nucleic Acids.Res.2003,31,3784.
December 17,2010;Revised:January 25,2011;Published on Web:March 18,2011.
Classification of Coenzyme-A Binding Proteins Based on Co-Factor Binding Modes
FAN Di§LIU Zhen-Ming§,*JIN Hong-Wei ZHANG Liang-Ren*
(State Key Laboratory of Natural and Biomimetic Drugs,School of Pharmaceutical Sciences,Peking University, Beijing 100191,P.R.China)
This study developed a mutual recognition of the proteins based on molecular classification, data mining strategies and the statistical clustering method,which was applied to study and classify clusters of coenzyme-A(CoA)binding proteins with their binding patterns extracted by using Pocket1.0 program.Several strategies have been evaluated for the accuracy of the system analysis and the two-step clustering method has been shown to be the best.The results revealed that the known CoA binding proteins can be clustered into three groups by using this approach.The designed classification coefficient was used effectively to identify the critical features for classification.The results show that both hydrogen bonds and hydrophobic interactions are important in all three clusters and that quite a few important residues related to biological activities are involved in the formation of hydrogen bonds.The classification of these interactions and the discovery of the characteristics and differences between the three clusters will have some utility for the design of specific agonists and antagonists.
Coenzyme-A;Protein classification;Binding mode;Cluster analysis;Pantetheine
O641
*Corresponding authors.ZHANG Liang-Ren,Email:liangren@bjmu.eud.cn;Tel:+86-10-82802567.LIU Zhen-Ming,Email:zmliu@bjmu.edu.cn; Tel:+86-10-82805514.
§These authors contributed equally to this work.
The project was supported by the Major National Science and Technology Program of Key Drug Scheme Funds,China(2009ZX09501-002)and National Natural Science Foundation of China(20802006).
重大新药创制国家科技重大专项(2009ZX09501-002)和国家自然科学基金(20802006)资助项目
