氨噻肟酸的合成
2011-07-27彭学伟
孙 健,彭学伟
(1.青岛农业大学化学与药学院,山东 青岛 266109;2.潍坊学院,山东 潍坊 261061)
氨噻肟酸是头孢菌素类抗生素的一种重要的侧链,可用于生产高效低毒、抗菌谱广的头孢类抗生素,如头孢曲松、头孢噻肟等,是很有发展前景的医药中间体[1,2]。
目前,氨噻肟酸多以乙酰乙酸乙酯为起始原料,经肟化、甲基化、氯化、环合、水解等步骤合成[3~5]。作者在此将肟化、甲基化合并为一步,简化了操作步骤,并对其它反应条件进行了优化,提高了收率,更加适合于实际应用。反应过程如下:
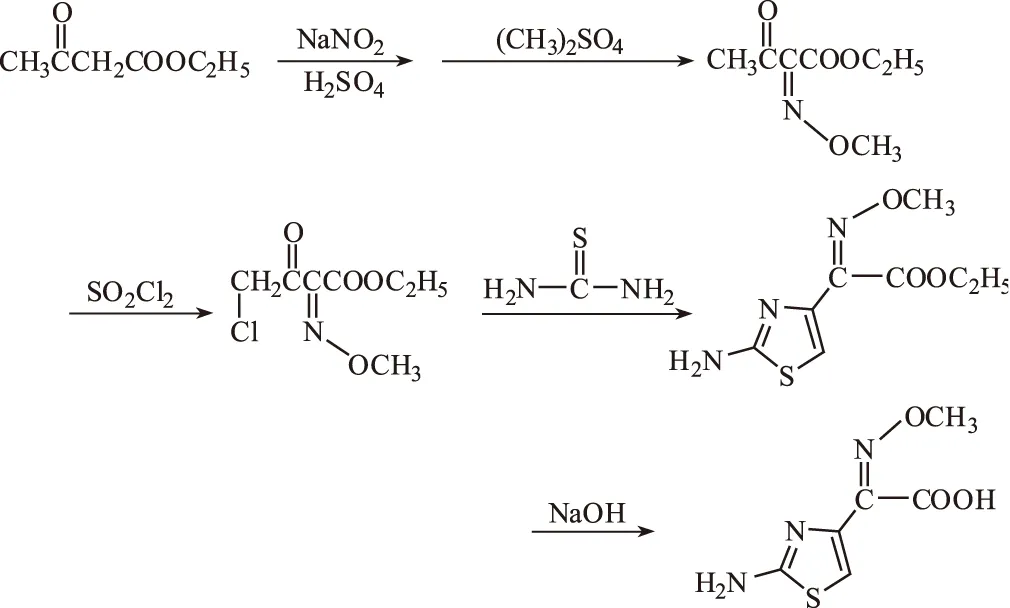
1 实验
1.1 试剂与仪器
所用试剂皆为市售分析纯,未做进一步纯化处理。
FX-90Q型核磁共振仪,日本电子公司;XT4型显微熔点仪,南京庚辰科学仪器公司;GF-254型硅胶板,烟台化工研究所。
1.2 方法
1.2.1 肟化、甲基化
向反应瓶中依次加入78 g(0.6 mol)乙酰乙酸乙酯、45 g(0.65 mol)亚硝酸钠、200 mL水。搅拌下滴加24 mL 98%的硫酸与100 mL水配制的溶液,温度控制在0~5 ℃。滴加完后,继续在此温度范围内搅拌2 h。反应毕,用200 mL二氯甲烷分3次萃取反应液。有机相以饱和食盐水洗涤,再向该萃取液中加入76 g(0.72 mol)Na2CO3、150 mL水、1 g四丁基溴化铵。室温下滴加90.7 g(0.72 mol)(CH3)2SO4,在30 ℃以下反应2 h。静置分层,分去水相。有机相以600 mL水分2次洗涤后,以无水硫酸钠干燥过夜。减压浓缩除去溶剂。得2-甲氧亚胺乙酰乙酸乙酯粗品98.5 g,低温保存备用。收率95%。
1.2.2 氯化[5]
取1.2.1产品69.2 g(0.4 mol)溶于40 mL CH2Cl2,放入反应瓶中,加入11 mLN,N-二甲基甲酰胺。室温下滴加64.8 g(0.48 mol)SO2Cl2。滴完后,升温到40 ℃,继续搅拌反应10 h。反应毕,缓缓加入60 mL水,静置,自然冷却分层。水相以10 mL CH2Cl2抽提,合并有机相,以无水硫酸钠干燥。减压浓缩除去溶剂,得淡黄绿色油状液体76.5 g。收率92.2%。
1.2.3 环合
取1.2.2产品62.3 g(0.3 mol)与60 mL乙醇配成混合溶液。取27.4 g(0.36 mol)硫脲、60 mL乙醇、120 mL水,放入反应瓶中,搅拌0.5 h后,冷却至15 ℃以下,滴加配制的混合溶液。加完后,继续搅拌反应3 h。整个过程控制温度低于15 ℃。反应毕,以氨水调pH值至7,继续搅拌反应45 min,抽滤。滤饼以水洗涤。50 ℃下真空干燥得类白色粉末状结晶46.1 g。收率60.8%。
1.2.4 水解
取1.2.3产品45.8 g(0.2 mol)悬浮于9 g NaOH与50 mL水配制的溶液中,加入50 mL乙醇,在40~45 ℃下搅拌反应1 h。体系完全透明后,将反应瓶置于冰浴中,待温度降到0 ℃左右,以冰乙酸调节pH值至6,维持此温度至结晶完全。快速抽滤。母液收集备用。滤饼以1∶1的乙醇与水的混合溶液洗涤,再用乙醚洗涤,常温真空干燥,得无色针状结晶28.3 g;母液真空浓缩,冷却结晶,同上处理得淡黄色粉末状结晶7.0 g。共得氨噻肟酸产品35.3 g。收率84.5%,总收率45%。
2 结果与讨论
2.1 合成路线的改进
由于氨噻肟酸的合成过程中,硫酸二甲酯与丙酮的用量大;反应每一步都要提纯,操作复杂;分离过程中会造成产品的损耗。因此,尝试将肟化与甲基化合并,并且采用四丁基溴化铵为相转移催化剂,在两相介质中进行甲基化,可以避免使用丙酮、甲醇等有机溶剂,减少硫酸二甲酯和碳酸钠的用量,确保甲基化进行得更充分、完全。不经分离,可以直接进行氯化反应,减少生产环节。同时,以无机酸代替有机酸,碱洗更容易操作,可降低成本、提高收率。
2.2 加料顺序的改进
研究中曾采用先氯化后肟化的加料顺序,发现收率较低,产品质量较差。这可能是由于乙酰乙酸乙酯在氯化时存在反应位置的选择性问题。α-位与γ-位均可能竞争氯化反应,影响到收率与产品质量。虽然在0 ℃以下反应可以避免α-位产物的生成,但是条件苛刻,不易操作。而先肟化时,α-位两个氢均参加反应,下一步不会再有副反应发生,因而收率较高,同时氯化可在室温或微热下进行,反应更加完全,操作更加容易。因此,采用先肟化后氯化的加料顺序。
2.3 环合过程的改进
2.3.1 控制温度的选择
氨噻肟酸乙酯存在顺反异构体。由于反式异构体没有药用价值,因此实验中应尽量避免反式异构体生成。这就要求控制好温度。从热力学上考虑,反式氨噻肟酸乙酯比顺式氨噻肟酸乙酯稳定;从动力学上考虑,顺式异构体更容易生成。因此,在低温下倾向于生成顺式产品(动力学控制),在高温下倾向于生成反式产品(热力学控制)。实验发现,当反应温度高于15 ℃时,将会得到顺式与反式产品的混合物;当体系温度低于15 ℃时,可以得到较纯的顺式产品。
2.3.2 溶剂的选择
不同溶剂对收率的影响见表1。
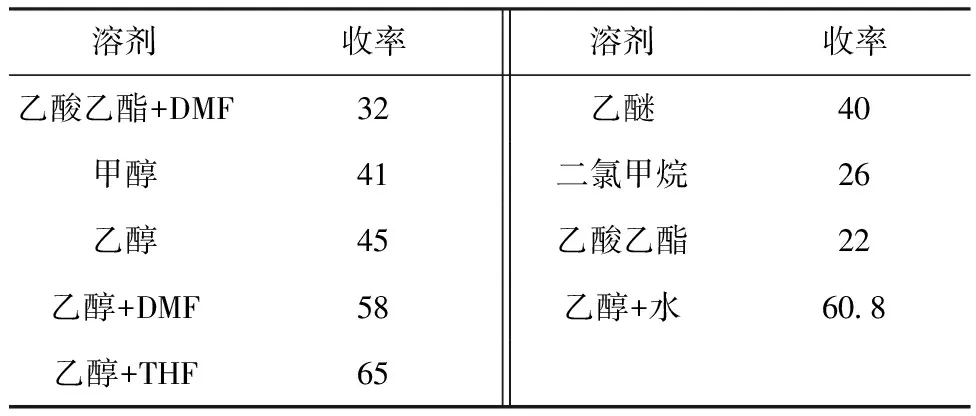
表1 不同溶剂对收率的影响/%
由表1可知,乙醇与四氢呋喃(THF)的混合溶液最利于反应的进行,收率最高;乙醇与DMF的混合溶液、乙醇与水的混合溶液收率也较高。综合考虑成本与产品的后处理问题,选择乙醇与水的混合溶液作为反应溶剂。
2.4 水解过程的改进
文献报道多在45 ℃左右进行水解反应,虽然反应速度快,大多在1 h内完成,但产品颜色较深,需用活性炭脱色。改在室温下进行反应后,虽然反应时间较长(一般需7 h),但无需用活性炭脱色,避免了脱色时的损失。
2.5 理化数据
2.5.1 熔点
中间体及目标产物的熔点如下:2-羟肟乙酰乙酸乙酯48~50 ℃;顺式氨噻肟酸乙酯158~161 ℃;顺式氨噻肟酸128~150 ℃。
2.5.21HNMR谱图解析
顺式氨噻肟酸乙酯:1HNMR(90 MHz,CDCl3),δ,ppm:1.22~1.38(t,3H,-CH3),3.93(s,3H,-OCH3),4.21~4.45(q,2H,-OCH2-),5.71(s,2H,-NH2),6.61(s,1H,噻唑5-H)。
顺式氨噻肟酸:1HNMR(90 MHz,DMSO-d6),δ,ppm:3.81(s,3H,-OCH3),5.00(s,5H,-NH2与水峰有重合),6.72(s,1H,噻唑5-H),7.00(s,1H,-OOH)。
3 结论
以乙酰乙酸乙酯为起始原料合成氨噻肟酸,对合成路线及反应条件进行了优化,目标产物总收率达到45%(文献值39%[5])。
[1] 于文洲,段大成.对我国β-内酰胺类抗生素发展的建议及十大城市用药情况分析[J].国外医药(抗生素分册),2001,22(4):145-150.
[2] Durckheimer W,Blumbach J,Lattrell R,et al.Recent developments in the field ofβ-lactam antibiotics[J].Angewandte Chemie(International Edition in English),1985,24(3):180-202.
[3] 葛洪玉,张静,方志杰.氨噻肟酸的生产及工艺评析[J].江苏化工,2005,33(1):5-6.
[4] 王之德.氨噻肟酸的制法[J].四川化工,1989,(4):41-43.
[5] 朱赛芬,严招春.氨噻肟酸生产技术与应用[J].化工生产与技术,2001,8(5):39-41.
