磷代谢与慢性肾脏病
2011-07-01王梦婧综述审校
王梦婧 综述 陈 靖 审校
·医学继续教育·
磷代谢与慢性肾脏病
王梦婧 综述 陈 靖 审校
磷是生命体中重要的元素之一,在细胞代谢和组织结构上起关键作用。磷以磷酸根形式存在,在细胞内组成细胞膜和遗传物质,参与细胞能量代谢及信号传导;在细胞外是骨无机质和牙结构的主要成分之一,部分则存在于血液循环,即临床可检出的血磷。血磷是机体磷代谢状况的直接反应,肠道吸收、肾脏排泄、组织利用以及一系列调节因子共同作用决定血磷的平衡。在慢性肾衰竭患者中,由于肾功能减退及身体内分泌功能的变化,这种平衡被打破而呈现高磷状态,已证实高磷血症除与骨代谢相关外,与心血管事件的发生和死亡率关系也甚密切。本文将围绕正常磷代谢、慢性肾脏病患者磷代谢异常以及高磷血症治疗的新进展进行综述。
慢性肾脏病 磷代谢 高磷血症
磷元素含量在人体内位列第六,85%存在于骨组织,14%存在于其他组织的细胞内,1%存在于细胞外液[1]。磷以磷酸根()形式存在于生物体,参与细胞功能的维持和代谢[2]。在细胞内,磷脂是细胞膜的重要组成部分,核苷酸参与DNA和RNA的形成,三磷酸腺苷与能量代谢密切相关,磷酸化则是细胞内信号转导的重要途径。在细胞外,羟磷灰石是骨无机质的主要成分,还有一部分磷在血浆中循环,即临床上所熟知的血磷。血磷指无机磷酸盐中所包含的磷,正常范围是2.5~4.5 mg/dl。在pH为7.4的血浆中,无机磷以HPO2-4∶H2PO-4=4∶1的形式存在。人体血浆磷酸盐的水平还存在节律变化,一般上午11:00最低,下午16:00升至平台期,午夜达高峰[3]。磷平衡对于生命体内环境稳态十分关键。本文将着重讨论生理情况下磷的代谢及慢性肾脏病(CKD)患者磷代谢异常的治疗。
正常磷代谢
磷的吸收和排泄 磷的吸收部位主要在空肠。食物中的肉类、鱼类、奶制品及添加剂等含磷量均很高,每日正常饮食大约含磷1~1.5g,主要是磷脂和磷酸脂,两者在消化液磷脂酶的作用下水解成无机磷酸盐,并以H2PO-4的形式被肠道吸收,吸收率是65%~70%。胃肠道中的阳离子如钙离子、镁离子、铝离子可结合磷酸盐生成不溶于水的化合物而影响磷吸收[2],未吸收的磷通过粪便排出。另外,胰腺和小肠每日主动分泌约200 mg磷,因此每日磷的净吸收量为500~850 mg。经肠道吸收后的磷进入细胞外液,进出组织、细胞和骨骼参与代谢[2]。机体磷的排泄主要通过肾脏,每日肾小球滤过的磷近5g,85%~95%的滤过磷能被近曲肾小管重吸收,远端肾小管重吸收少量,肾脏重吸收磷的总量根据身体需要而改变[4]。
钠磷协同转运子(Na-Pi cotransporter,NPC)是决定小肠和肾脏磷转运的共同细胞通道,分三种亚型,即Ⅰ型、Ⅱ型和Ⅲ型(图1)。NPC-I主要存在近端肾小管,介导钠磷协同转运、氯离子转运[5],也有可能参与有机阴离子的排泄。NPC-Ⅱ分为a、b、c三种,Ⅱa型表达于近端肾小管的刷状缘,Ⅱb型表达于近端小肠的刷状缘,两者均为特异性的钠离子依赖的磷吸收通道,是生理调节的靶目标[6],Ⅱc型也表达于近端肾小管刷状缘,存在于肾脏表层或中间皮质的肾单位中,可能与新生儿时期的磷平衡有关[7]。NPC-Ⅲ几乎存在于机体所有的组织和细胞中,起着“管家”的作用,保证细胞代谢所需要的磷不外流[4,6]。另外,磷在肠道有很少一部分是通过上皮细胞间隙进入细胞外液,而磷从细胞基侧膜转运至细胞外的机制目前还不清楚。
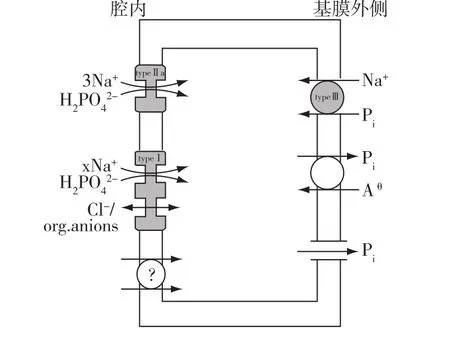
图1 钠磷转运子Ⅰ型、Ⅱ型和Ⅲ型转运图[5]
影响因素
饮食中的磷含量 研究证实低磷饮食会导致肾小管100%重吸收磷,高磷饮食则抑制磷的重吸收,该调节机制是通过改变肾小管上皮细胞NPC数量,且不受甲状旁腺激素(PTH)和维生素D等激素水平变化的影响[8]。
甲状旁腺激素(PTH) PTH通过调节钠磷转运子的数量来改变肾小管对磷的重吸收,急性反应可能与钠磷转运子内吞、溶酶体降解有关,慢性反应则与抑制肾小管NPC基因转录和蛋白合成有关[9]。PTH还可通过刺激肾脏1α羟化酶,促进1,25羟维生素D3[1,25(OH)2D3]的合成而间接增强钙磷的吸收[1]。
1,25羟维生素D31,25(OH)2D3可抑制肾脏对磷的排泄,但更重要的是增加肠道对钙磷的吸收[1]。维生素D作用下的磷酸转运系统在小肠是独立的、与钙转运完全无关的主动运输过程,并依赖特异的载体磷酸转移蛋白[10]。
成纤维细胞生长因子23(fibroblast growth factor-23,FGF-23) FGF-23是多肽激素成纤维细胞生长因子家族成员,主要由成骨细胞分泌,肾脏是其主要的靶器官。研究证实 FGF-23能抑制NPC-Ⅱa、Ⅱc型的表达与合成,促进磷在肾脏的排泄;同时抑制肠道NPC-Ⅱb型的活性,减少Ⅱb型数量,抑制肠道磷的吸收[11]。FGF-23还能抑制1α羟化酶的基因转录,减少1,25(OH)2D3的合成,同时增加24羟化酶的合成,促进1,25(OH)2D3降解,间接影响磷代谢[12]。Klotho蛋白是一种与衰老相关的因子,与FGFR1c协同作用构成了FGF-23受体复合物,主要表达于远端肾小管,在甲状旁腺、垂体和脉络丛也可见到该复合体的表达。当饮食中磷含量增加时,体内FGF-23的水平增加。血清全段甲状旁腺素(iPTH)、血钙和1,25(OH)2D3可能也参与了FGF-23分泌的调节[13]。
胰岛素 DeFronzo等[14]证实在高胰岛素血症的狗中磷的重吸收量增加,相反给予生长抑素降低血浆胰岛素水平后磷的排泄增加,调节机制可能与肾小管NPC有关。
生长激素 在近端肾小管的基膜侧存在生长激素受体,通过激活磷脂酶C的信号途径产生细胞内效应,增强NPC对磷的重吸收[5]。生长激素还可促进肾脏合成胰岛素样生长因子1刺激NPC的转运功能,这种效应在15 min时即可出现,5h达到高峰[1]。
多巴胺 研究发现将左旋多巴转换成多巴胺的酶只存在于近端肾小管中,在体外培养的肾脏细胞中,多巴胺能显著减少磷的转运,通过多巴胺受体促使钠磷转运子内吞,因此多巴胺有可能是由近端肾小管旁分泌产生、局部调节磷代谢的物质[15]。
其他 肾小管管腔内碱性环境可以刺激钠磷转运子的转运,而慢性代酸或呼酸则抑制NPC转运。此外,肾上腺皮质激素、血容量的改变等都能引起磷代谢的改变[5]。
CKD患者的磷代谢
CKD患者矿物质代谢可发生明显改变,常出现高磷血症、高 PTH血症、低钙血症和低1,25(OH)2D3血症。在肾功能减退初期,磷潴留可刺激PTH分泌增多,抑制肾小管对磷的重吸收,临床上并不出现高磷血症。当肾功能进行性减退[肾小球滤过率(GFR)<20~30 ml/min]时,尽管血PTH升高,但有效肾单位减少,肾小管对PTH的反应能力下降,肾脏对磷的排泄发生障碍,磷在体内积蓄,出现高磷血症。同时,肾脏1α羟化酶减少致使1,25(OH)2D3的合成降低,小肠对钙的吸收减少,而骨骼对PTH的抵抗更加剧了低钙血症的形成。低钙血症、低 1,25(OH)2D3血症、甲状旁腺对1,25(OH)2D3抑制的敏感度降低以及高磷血症均可导致高PTH血症的形成。
越来越多证据表明,FGF-23也参与CKD患者血磷调节,且与低1,25(OH)2D3,继发甲状旁腺功能亢进有关。早在高磷血症出现之前,CKD患者就有血FGF-23的升高,随着GFR的下降,FGF-23进一步升高。当 GFR>80 m l/min时,血清1,25(OH)2D3水平和肾小管磷的最大重吸收率与血清FGF-23呈负相关;当GFR<30 m l/min时,血FGF-23水平较高,但与尿磷清除无明显相关性[16]。由于FGF-23能够抑制1α羟化酶的活性,它可能是1,25(OH)2D3降低的原因,也可能是继发性甲状旁腺功能亢进的早期促使因子[17]。关于FGF-23升高的原因尚不明确,可能与肾脏清除减少、长期维生素D治疗及高血磷等诸多因素有关。大多数透析患者血清FGF-23的浓度超过正常人的100多倍,是血液透析(HD)患者心血管事件及死亡率的独立危险因子[18]。
高磷血症的危害
研究证实血磷升高可以:(1)导致血清中矿物质代谢失衡,进而降低了机体调节矿物质处于溶解状态的能力,使矿物质沉积的阈值降低,同时增高钙磷乘积,形成不溶性磷酸钙,易于在心血管和软组织内沉积[19];(2)干扰PTH和1,25(OH)2D3代谢,导致肾性骨营养不良[20];(3)抑制肾脏1α羟化酶的活性,降低血1,25(OH)2D3水平,诱发低钙血症,刺激甲状旁腺细胞增生,促进甲状旁腺功能亢进。除了这种负反馈调节外,高血磷也可直接刺激甲状旁腺细胞增生[21]。继发性甲状旁腺功能亢进又加速骨盐溶解而释放更多钙、磷,从而加重高磷、低钙血症和1,25(OH)2D3的缺乏,导致钙磷代谢紊乱的恶性循环。
在维持性血液透析(MHD)患者的调查中发现,心、脑血管疾病是其主要死因,而心血管系统的钙化是造成心血管疾病高发生率和高死亡率的重要因素之一。其中血磷水平在心血管钙化的发生发展中比血钙水平更为重要,是预测冠状动脉钙化、颈动脉内膜中膜增厚和心脏瓣膜钙化的独立危险因素[22]。此外,高血磷与MHD患者的死亡率密切相关。Block等[23]回顾性分析了6 407例HD患者发现,血磷每上升0.32 mmol/L相对死亡危险性增加6%;校正了合并症、尿素清除指数(Kt/V)、营养参数和顺应性差等指标后,血磷>2.10 mmol/L的患者与血磷0.78~2.10 mmol/L患者相比,死亡危险性增加27%。高磷血症促进血管钙化的机制尚不完全明确,研究表明,除了钙盐在骨外软组织异常沉积外,高血磷还可直接诱导血管平滑肌细胞骨桥接素和碱性磷酸酶的表达而促进血管钙化;高血磷通过NaPi-Ⅲ的作用,导致细胞内磷升高,诱导血管平滑肌细胞转化为类成骨细胞,表达参与血管钙化的选择性骨相关蛋白及骨特异性蛋白(如Osteocalcin和Cbfa-1),促进血管钙化;磷还可直接诱导血管中层平滑肌细胞凋亡,继而发生血管壁钙化[24]。
CKD患者高磷血症的治疗进展
早在CKD 2期的患者已出现磷潴留,且成为继发性甲状旁腺功能亢进的发病基础;CKD 3期时,血浆PTH水平开始升高,通常此时血磷和血钙的水平仍属正常范围;CKD 4~5期时,血磷水平出现升高。因此,要控制高磷血症,必须严密监测CKD患者的血磷水平。K/DIGO指南建议CKD 3期患者,血磷每6~12个月监测一次;CKD 4期患者,每3~6个月监测一次;CKD 5期患者,每1~3个月监测一次。当生化指标出现异常或开始肾性骨病治疗后,应适度增加监测频率[25]。目前控制高磷的方法主要为以下几个方面:
重视限磷饮食 一般提倡患者在CKD 2期开始限磷饮食,但依从性很差,国外资料表明仅31%~35%患者能够配合治疗。当患者进入CKD 3期甚至CKD 4、CKD 5期时,限磷饮食比较容易实施。然而,为避免MHD患者出现营养不良,K/DOQI指南推荐蛋白质摄入量应>1.2 g/(kg·d),相应磷的摄入量就达到1 000~1 400 mg/d。即使是最严格的限磷饮食,每天仍有600 mg的磷进入体内[5]。最近的一项研究表明,降低HD患者蛋白摄入量至0.8 g/(kg·d),同时补充酮酸类制剂,患者的营养状态仍然维持良好,且血磷降至正常[26]。因此对于此类患者可采用低蛋白饮食或选用含磷相对少的蛋白质,同时补充酮酸类制剂的方法。
磷结合剂 当HD患者饮食控制磷摄入难以达标时,就需要磷结合剂来减少磷从肠道的吸收。铝盐因有严重的不良反应(如骨软化病、脑病、贫血等)已不常规使用[27]。碳酸钙是目前中国市场上使用最广泛的磷结合剂,但该药物有增加血钙的风险,在使用活性维生素D治疗时尤为显著。然而,当患者存在反复出现高钙血症、血管钙化、低动力型骨病及低PTH血症时,不推荐使用含钙的磷结合剂[28]。醋酸钙比碳酸钙对血钙水平影响相对小,但醋酸钙的胃肠道反应发生频繁,患者的依从性不如碳酸钙。新型磷结合剂盐酸司维拉姆是一种阳离子聚合物。盐酸司维拉姆的降磷效果不仅与含钙的磷结合剂相同,且有降低血脂、减少炎症、降低尿酸、减少氧化应激及改善骨病等作用,不易造成高钙血症。一项荟萃分析表明与钙磷结合剂比较,盐酸司维拉姆尽管未改善MHD患者的心血管钙化程度,但是能降低C反应蛋白、碱性磷酸酶及PTH水平[29]。碳酸镧是另一种新型的磷结合剂,在胃或十二指肠和空肠均能与磷高效结合,形成不溶性的、不易被消化道吸收的镧盐,然而也有文献报道吸收的碳酸镧能沉积于肝脏、骨骼、肌肉、肾脏、大脑等,并且为时间依赖性,因此长期服用碳酸镧的安全性仍未得到评价[27]。一项关于碳酸镧和碳酸钙的研究表明在达到同等降磷效果的基础上,碳酸镧和碳酸钙引起高钙血症的比例分别是0.4%和20%,并且该药的耐受性较好[16]。聚苯乙烯磺酸镧是最近新推出的磷结合剂,其与磷的亲和力在pH 5~7时,比碳酸镧高5倍,是目前最好的磷酸盐结合剂[30]。近来有研究发现烟胺能抑制动物小肠上的NPC,从而降低血清磷水平。Sampathkumar等[31]的前瞻性研究证明,烟酸能降低HD患者血清磷水平。因此,烟酸可能成为治疗伴有高脂血症的终末期肾病患者高磷血症的理想药物。另外,有临床试验发现,健康志愿者服用考来替兰后,粪便中磷排泄量明显增加,可见考来替兰可能具有磷结合能力。Locatelli等[32]为评估考来替兰作为磷结合剂的有效性和安全性进行了一项随机双盲安慰剂对照的前瞻性研究发现,考来替兰组血清磷水平较安慰剂组显著降低,其他的磷结合剂有镁盐、含铁复合物等。镁盐的磷结合能力较弱,且有很强的致泻作用,治疗中可诱发高镁血症,因而目前主要应用于伴有便秘的患者。含铁复合物可以促进消化道排磷、降低血磷而不增加血清钙和钙磷乘积,同时可以纠正缺铁性贫血,但多数铁剂的磷结合效力较低,故未能在临床推广应用。
透析 当患者进入CKD 5期后,透析成为清除磷的方法之一。研究证明,透析前血磷越高、透析膜面积越大、透析频率越大、透析时间越长、采用血液滤过、透析前PTH水平越高、透析磷的清除越多。透析膜材料、透析缓冲液的种类、血流速度、Kt/V、透析液流速并不能明显影响透析磷的清除。而年龄越大、血红蛋白越高,透析磷清除越少[33]。由于磷从细胞内释放入血的速度远远慢于透析过程中磷的清除速度,因此常规HD对磷的清除是不充分的,平均4h仅能清除700~1 000 mg磷[33],每周清除2.1~3g磷。持续性不卧床腹膜透析每天排磷仅为310~320 mg/d,每周清除2.1g左右。假设透析患者每日摄入的磷为800 mg,那么每周摄入的磷高达5.6g,因此透析清除的磷远不及摄入的磷总量,透析效果十分有限。每日短时透析(short daily HD,SDHD,5~6次/周,1.5~2.5 h/次)或每天夜间透析(daily nocturnal HD,DNHD,5~7晚/周,6~8 h/晚)均能良好控制血磷到目标水平[34],减少左心室肥大、减少高血压用药、改善矿物质代谢及生活质量,但这些透析方法的实施亦受到多重因素的限制。
小结:正常磷代谢是维持机体一系列生理功能的重要因素,CKD患者容易出现磷潴留,高磷血症与各种并发症及死亡率密切相关。对于高磷血症的治疗,不仅仅在于磷水平的控制,必须同时控制血钙、血PTH及血1,25(OH)2D3水平。根据最新的K/DIGO指南,建议:CKD 3~4期的患者血钙磷水平应控制在正常范围内(血钙:8.5~10.5 mg/dl,血磷2.5~4.5 mg/dl),CKD 5期患者血磷应尽量接近正常范围(较K/DOQI推荐的范围更加严格);建议对个体的血钙磷水平共同评估指导临床治疗,而非钙磷乘积。CKD 3~4期患者PTH的理想水平尚未确定;CKD 5期患者,建议PTH水平维持在正常上限的2~9倍。CKD 3~5期患者,根据基线维生素D水平和治疗干预措施决定复查维生素D的频率,建议按照普通人群的治疗方案纠正CKD患者的维生素D缺乏和不足。所有治疗方案的确定应基于病情的动态变化,而非某一项监测数据[25]。
1 Caverzasio J,Bonjour JP.Mechanism of rapid phosphate(Pi)transport adaptation to a single low Pimeal in rat renal brush border membrane.Pflugers Arch,1985,404(3):227-231.
2 Kestenbaum B.Phosphatemetabolism in the setting of chronic kidney disease:significance and recommendations for treatment.Semin Dial,2007,20(4):286-294.
3 Portale AA,Halloran BP,Morris RC Jr.Dietary intake of phosphorus modulates the circadian rhythm in serum concentration of phosphorus. Implications for the renal production of 1,25-dihydroxyvitamin D.J Clin Invest,1987,80(4):1147-1154.
4 Reshkin SJ,Forgo J,Biber J,et al.Functional asymmetry of phosphate transport and its regulation in opossum kidney cells:phosphate“adaptation”.Pflugers Arch,1991,419(3-4):256-262.
5 Murer H,Hernando N,Forster I,et al.Proximal tubular phosphate reabsorption:molecularmechanisms.Physiol Rev,2000,80(4):1373 -1409.
6 Murer H,Hernando N,Forster L,et al.Molecular mechanisms in proximal tubular and small intestinal phosphate reabsorption(plenary lecture).Mol Membr Biol,2001,18(1):3-11.
7 Ohkido I,Segawa H,Yanagida R,et al.Cloning,gene structure and dietary regulation of the type-Ⅱc Na/Pi cotransporter in the mouse kidney.Pflugers Arch,2003,446(1):106-115.
8 Barac-Nieto M,Alfred M,Spitzer A.Phosphate depletion in opossum kidney cells:apical but not basolateral or transepithelial adaptions of Pi transport.Exp Nephrol,2001,9(4):258-264.
9 Bacic D,Lehir M,Biber J,et al.The renal Na+/phosphate cotransporter NaPi-Ⅱa is internalized via the receptor-mediated endocytic route in response to parathyroid hormone.Kidney Int,2006,69(3):495-503.
10付强,刘 源.钙、磷与维生素D对动物骨代谢的影响研究进展.中国比较医学杂志,2006,16(8):502-504.
11 Saito H,Kusano K,KinosakiM,et al.Human fibroblast growth factor-23 mutants suppress Na+-dependent phosphate co-transport activity and 1alpha,25-dihydroxyvitamin D3 production.JBiol Chem,2003,278(4):2206-2211.
12 Berndt TJ,Schiavi S,Kumar R.“Phosphatonins”and the regulation of phosphorus homeostasis.Am JPhysiol Renal Physiol,2005,289(6):F1170-1182.
13 Urakawa I,Yamazaki Y,Shimada T,et al.Klotho converts canonical FGF receptor into a specific receptor for FGF23.Nature,2006,444(7120):770-774.
14 Hammerman MR,Rogers S,Hansen VA,et al.Insulin stimulates Pi transport in brush border vesicles from proximal tubular segments.Am JPhysiol,1984,247(5 Pt1):E616-624.
15 Berndt TJ,MacDonald A,Walikonis R,et al.Excretion of catecholamines and metabolites in response to increased dietary phosphate intake.JLab Clin Med,1993,122(1):80-84.
16 Shigematsu T,Kazama JJ,Yamashita T,et al.Possible involvement of circulating fibroblast growth factor23 in the developmentof secondary hyperparathyroidism associated with renal insufficiency.Am JKidney Dis,2004,44(2):250-256.
17 Stubbs J,Liu S,Quarles LD.Role of fibroblast growth factor 23 in phosphate homeostasisand pathogenesisof disorderedmineralmetabolism in chronic kidney disease.Semin Dial,2007,20(4):302-308.
18 Gutiérrez OM,Mannstadt M,Isakova T,et al.Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis.N Engl J Med,2008,359(6):584-592.
19苗 华,潘明明.慢性肾衰竭高磷血症研究及治疗进展.中国血液净化,2007,6(9):500-502.
20 Arcidiacono T,Paloschi V,Rainone F,et al.Renal osteodystrophy and vascular calcification.JEndocrinol Invest,2009,32(4 Suppl):21-26.
21 Zhang Q,Qiu J,Li H,et al.Cyclooxygenase 2 promotes parathyroid hyperplasia in ESRD.JAm Soc Nephrol,2011,22(4):664-672.
22 Ishimura E,Taniwaki H,Tabata T,et al.Cross-sectional association of serum phosphate with carotid intima-medial thickness in hemodialysis patients.Am JKidney Dis,2005,45(5):859-865.
23 Block GA,Hulbert-Shearon TE,Levin NW,et al.Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients:a national study.Am J Kidney Dis,1998,31(4):607-617.
24 Raggi P,Kleerekoper M.Contribution of bone and mineral abnormalities to cardiovascular disease in patientswith chronic kidney disease.Clin JAm Soc Nephrol,2008,3(3):836-843.
25 Kidney Disease:Improving Global Outcomes(KDIGO)CKD-MBD Work Group.KDIGO clinical practice guideline for the diagnosis,evaluation,prevention,and treatment of Chronic Kidney Disease-Mineral and Bone Disorder(CKD-MBD).Kidney Int Suppl,2009,(113):S1-130.
26 Li H,Long Q,Shao C,et al.Effect of short-term low-protein diet supplemented with keto acids on hyperphosphatemia in maintenance hemodialysis patients.Blood Purif,2011,31(1-3):33-40.
27 Schucker JJ,Ward KE.Hyperphosphatemia and phosphate binders. Am JHealth Syst Pharm,2005,62(22):2355-2361.
28 Kovesdy CP.How KDIGOwill(orwill not)influence themanagementof hyperphosphatemia.Semin Dial,2011,24(1):35-36.
29 Zhang Q,Li M,Lu Y,et al.Meta-analysis comparing sevelamer and calcium-based phosphate binders on cardiovascular calcification in hemodialysis patients.Nephron Clin Pract,2010,115(4):c259-267.
30袁发焕,杜 翔.高磷血症的危害及其防治.中国中西医结合肾病杂志,2010,11(10):847-849.
31 Sampathkumar K,Selvam M,Sooraj YS,et al.Extended release nicotinic acid-a novel oral agent for phosphate control.Int Urol Nephrol,2006,38(1):171-174.
32 Locatelli F,Dimkovic N,Pontoriero G,et al.Effect of MCI-196 on serum phosphate and cholesterol levels in haemodialysis patients with hyperphosphataemia:a double-blind,randomized,placebo-controlled study.Nephrol Dial Transplant,2010,25(2):574-581.
33 Kuhlmann MK.Management of hyperphosphatemia.Hemodial Int,2006,10(4):338-345.
34 Lindsay RM,Alhejaili F,Nesrallah G,et al.Calcium and phosphate balance with quotidian hemodialysis.Am JKidney Dis,2003,42(1 Suppl):24-29.
Phosphorusmetabolism and chronic kidney disease
WANGMeng-jing,CHEN Jing
Division of Nephrology,Huashan Hospital,Shanghai Medical College,Fudan University,Shanghai200040,China
Phosphorus is an important element in the life of the body in cellular metabolism and structure maintenance.Serum phosphorus is a direct reflection of phosphorusmetabolism.The balance of phosphorus is decided by intestine absorption,kidney excretion,and body usage aswell as a series of regulatory factors.In patientswith chronic renal failure,this balance is broken.Patients often show high phosphorus status due to renal dysfunction and physical changes in endocrine function.Hyperphosphatemia has been proved to be related with renal osteodystrophy,vascular calcification,cardiovascular events incidence and mortality.Sodium phosphate co-transporter is the common channel to determine the phosphate transport in small intestine and renal tubular epithelial cells.It is divided into three subtypes.The impact factors of sodium phosphate co-transporter include diet,PTH,VitD3,FGF-23 and so on.The current means of control of hyperphosphatemia primarily are dialysis,diet restriction,and usage of phosphate binders.At the same time,the levels of serum calcium,PTH,and 1,25(OH)2D3are required to control.The latest K/DIGO guidelines recommend that in CKD3~4,serum calcium and phosphorus levels should be controlled within the normal range(calcium:8.5~10.5 mg/dl,phosphorus 2.5~4.5 mg/dl),while to CKD5 patients,serum phosphate should be as close to normal range.All treatment programs are determined according to the dynamic changes of patients'conditions,rather than a particularmonitoring data.
chronic kidney disease phosphorusmetabolism hyperphosphatemia
2011-08-20
(本文编辑 心 平)
复旦大学附属华山医院肾病科复旦大学肾脏病研究所(上海,200040)
