兼具抗氧化和γ⁃氨基丁酸增强活性的多功能抗脑卒中化合物
2022-05-20郑礼平王玉婷倪罗番陈盼盼秦亚娟厉廷有
郑礼平,王玉婷,倪罗番,陈盼盼,秦亚娟,厉廷有
南京医科大学药学院,江苏 南京 211166
脑卒中是世界上导致人类残疾和死亡的主要原因[1]。依达拉奉是一个低分子量的抗氧化药物,可以清除多种自由基,临床上主要用于脑卒中和肌萎缩性脊髓侧索硬化症(amyotrophic lateral sclerosi,ALS)的治疗[2-3]。脑卒中发生时,随着氧气和能量物质的耗竭,发生一系列生理生化过程,如钙离子的流入、激酶的激活、细胞膜的去极化、自由基的产生、凋亡信号通路的激活等,最终导致神经细胞死亡[4]。因此,只针对其中一个环节的药物其治疗作用有限,或者在动物模型中有效而难以临床转化[5]。
研究表明,发生脑卒中和随后的再灌注时,谷氨酸大量释放,通过N⁃甲基⁃D⁃天冬氨酸(N⁃methyl⁃D⁃aspartic acid,NMDA)受体介导的神经兴奋性毒性是神经细胞损伤的重要因素[5]。但NMDA受体拮抗剂由于其不良反应众多不易成药。γ⁃氨基丁酸(GABA)受体系统是抑制性神经系统,对NMDA 受体系统的兴奋性具有抑制作用。因此,GABA 受体激动剂具有神经保护作用。曾有GABA受体激动剂氯美噻唑进入三期临床,但由于仅作用于信号系统中的一个环节,治疗效果不显著,最终没有成为治疗药物[7]。丙泊酚是临床上使用的麻醉药物[8],是GABA 受体的增强剂和激动剂。研究表明,丙泊酚具有神经保护作用,其药理学机制基于其GABA 受体激动活性和其自身的抗氧化作用[9]。
根据分子骨架融合原理[10],结合丙泊酚和依达拉奉的结构特点,本文概念验证性地设计并合成了2 个依达拉奉类似物,希望其同时具有抗氧化活性和GABA活性。活性研究结果表明,化合物6b兼具抗氧化活性和GABA 活性,并显示出比依达拉奉更优的神经保护活性。
1 材料和方法
1.1 材料
1H NMR 用日本JEOL 的400 MHz 核磁共振仪测定,四甲基硅烷(TMS)为内标;质谱由美国Agilent⁃6410 LC/MS质谱仪测定;薄层层析用硅胶板为青岛化工研究所生产,用荧光、碘或硫酸显色;全自动多功能酶标仪(FLUOstar Omega,BMG LABTECH公司,德国);气浴恒温摇床(CHA⁃S THZ⁃82 ZD⁃85,江苏正基仪器有限公司);数码相机(W800,索尼株式会社,日本)。化学试剂除特殊说明外均为市售试剂。本研究所有动物实验操作经由南京医科大学实验动物福利伦理审查委员会许可(IACUC⁃1812012)。
1.2 方法
1.2.1 化合物的合成
化合物的合成方法如图1所示。烷基取代的酚经硝化得对位取代的化合物1,酚羟基乙酰化得化合物2,2 经H2/Pt 还原得化合物3,3 经重氮化反应并还原得中间体4,4直接用于和乙酰乙酸乙酯缩合得化合物5,5在碱性条件下脱乙酰基得终产物6。
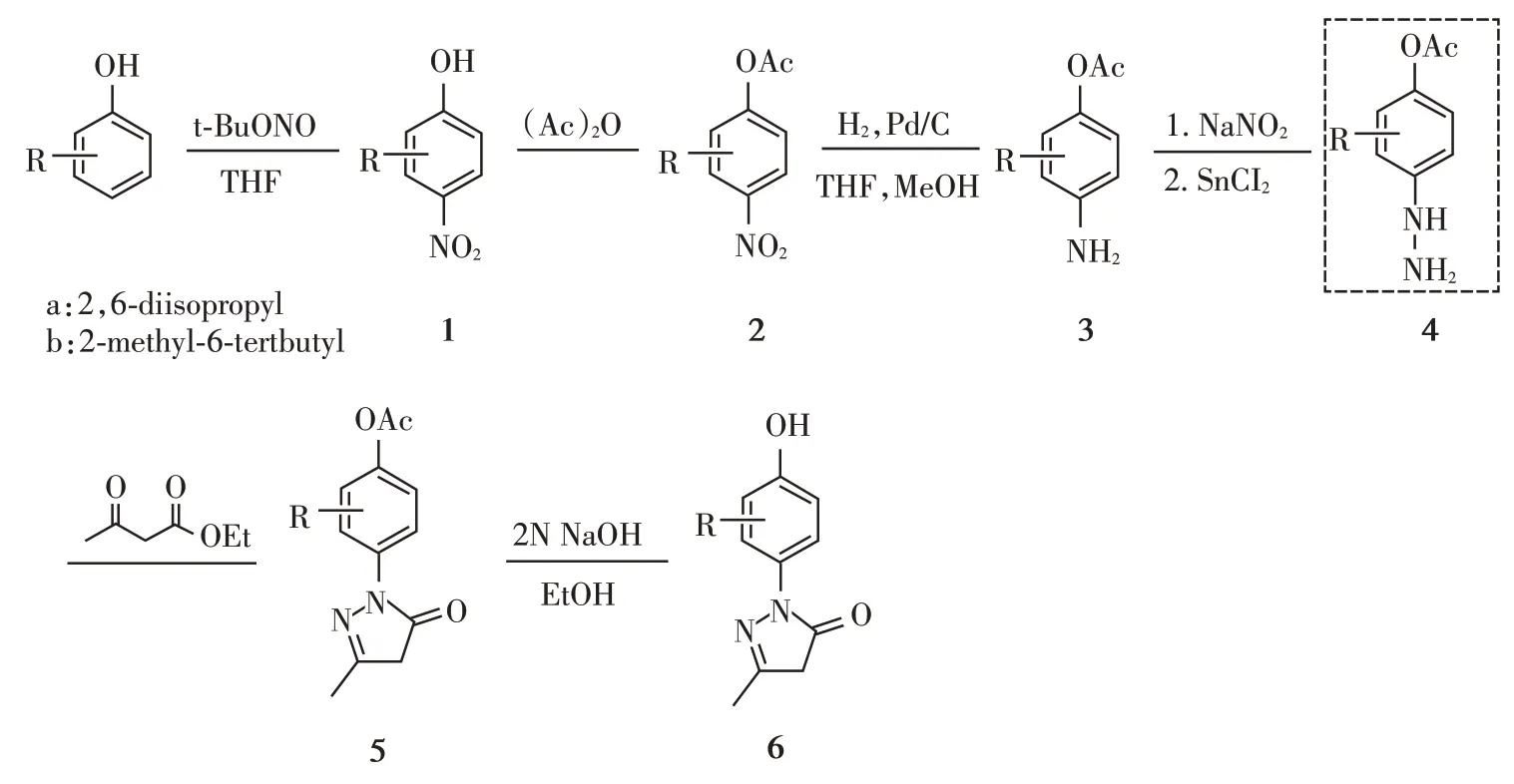
图1 化合物的合成方法Figure 1 Synthetic scheme for the compounds
1.2.1.1 2,6⁃二异丙基⁃4⁃硝基苯酚(1a)的合成
烧瓶中加入四氢呋喃100 mL,丙泊酚3.56 g(20 mmol),搅拌10 min。取亚硝酸叔丁酯4.8 mL(40 mmol),在冰浴条件下,缓慢滴加至上述溶液中,滴毕,40 ℃反应过夜。反应停止后,旋干溶液,得到棕褐色固体,加入150 mL石油醚,研磨至细小固体,超声5 min,过滤,得到橘色粗品。粗品进一步经过硅胶柱层析(PE∶EA=20∶1,Rf=0.3)纯化可得淡黄白色固体1.2 g,产率27%。1H NMR(400 MHz,CDCl3)δ:7.98(s,2H,arom),5.51(s,1H,OH),3.16(m,2H,CH(CH3)2),1.30(d,J=6.8Hz,12H,CH3).MS(ESI,m/z):222.1[M⁃H]-。
1.2.1.2 2⁃甲基⁃6⁃叔丁基⁃4⁃硝基苯酚(1b)的合成
合成方法同化合物1a。2⁃甲基⁃6⁃叔丁基苯酚3.28 g(20 mmol),亚硝酸叔丁酯4.8 mL(40 mmol),四氢呋喃100 mL,反应处理后,得到淡黄白色固体1b 1.5 g,Rf=0.2(PE∶EA=20∶1),产率36%。1H NMR(400 MHz,CDCl3)δ:8.09(d,J=2.8 Hz,1H,arom),7.95(d,J=2.8 Hz,1H,arom),5.45(s,1H,OH),2.32(s,3H,arom CH3),1.43(s,9H,C(CH3)3).MS(ESI,m/z):210.1[M+H]+。
1.2.1.3 2,6⁃二异丙基⁃4⁃硝基乙酰苯酚(2a)的合成
在125 mL 耐压瓶中依次加入1a 2 g(9 mmol),10 mL 醋酸酐,氩气置换后160 ℃反应过夜。反应停止后,加入乙酸乙酯100 mL,分别用300 mL 水洗涤有机层5次,饱和食盐水洗涤2次,无水硫酸钠充分干燥。过滤,旋干得产物2.3 g,产率96%。1H NMR(400 MHz,CDCl3)δ:7.98(s,2H,arom),2.95(m,2H,CH(CH3)2),2.39(s,3H,CH3),1.24(d,J=6.8 Hz,12H,CH3).
1.2.1.4 2⁃甲基⁃6⁃叔丁基⁃4⁃硝基乙酰苯酚(2b)的合成
合成方法同化合物2a。从2a 1.5 g(7.2 mmol)出发,反应、处理后得到淡黄色固体2b 1.75 g,Rf=0.8(PE∶EA=20∶1),产率97%。1H NMR(400 MHz,CDCl3)δ:8.16(d,J=2.8 Hz 1H,arom),8.01(d,J=2.8Hz,1H,arom),2.4(s,3H,arom CH3),2.22(s,3H,CH3CO),1.39(s,9H,C(CH3)3)。
1.2.1.5 2,6⁃二异丙基⁃4⁃氨基乙酰苯酚(3a)的合成
2a 2.3 g 溶解于25 mL 四氢呋喃和25 mL 甲醇中,加入10%钯碳0.25 g,氢气反应过夜。反应停止后,过滤除去钯碳,滤液旋干,硅胶柱层析(PE∶EA=5∶1,Rf=0.4)得淡黄白色固体3a 2 g,产率98%。1H NMR(400 MHz,DMSO⁃d6)δ:6.3(s,2H,arom),4.85(s,2H,NH2),2.68(m,2H,CH(CH3)2),2.22(s,3H,CH3),1.03(d,J=6.8 Hz,12H,CH3).MS(ESI,m/z):258.2[M+Na]+。
1.2.1.6 2⁃甲基⁃6⁃叔丁基⁃4⁃氨基乙酰苯酚(3b)的合成
合成方法同化合物3a。2b 1.75 g(7 mmol),10%钯碳0.2 g,四氢呋喃25 mL,甲醇25 mL。经还原处理后,得到淡黄色固体3b 1.5 g,Rf=0.5(PE∶EA=5∶1),产率97%。1H NMR(400 MHz,DMSO⁃d6)δ:6.43(d,J=2.8 Hz,2H,arom),6.28(d,J=2.8Hz,2H,arom),4.86(s,2H,NH2),2.26(s,3H,arom CH3),1.99(s,9H,C(CH3)3),1.87(s,3H,CH3CO).MS(ESI,m/z):221.1[M+H]+。
1.2.1.7 3,5⁃二异丙基⁃4⁃乙酰氧基苯肼(4a)的合成
3a 2.0 g(8.5 mmol)溶于6 mL 醋酸(102 mmol)和17 mL 浓盐酸(204 mmol)中,在冰浴条件下缓慢滴加亚硝酸钠(0.88 g,12.7 mmol)的水溶液(3.7 mL,204 mmol),维持温度0~5 ℃。反应1 h后,在氩气氛围中,在上述重氮盐中缓慢滴加二氯亚锡二水合物(5.75 g,25.5 mmol)的浓盐酸溶液(4.25 mL,51 mmol),维持温度0~5 ℃。反应6 h 后,2 mol/L 氢氧化钠调节pH=9,析出大量氢氧化亚锡沉淀,通过硅藻土过滤,滤液用乙酸乙酯萃取3 次,硅藻土用乙酸乙酯洗涤3次,合并所得有机层,饱和食盐水洗涤2次,无水硫酸钠充分干燥,过滤旋干得橘色油状液4a。苯肼类化合物易氧化变质,因此,不经分离纯化直接用于下一步合成。
1.2.1.8 3’,5’⁃二异丙基⁃4’⁃乙酰氧基依达拉奉(5a)的合成
上述得到的4a用15 mL 冰醋酸溶解,冷水浴条件下搅拌10 min,缓慢滴加乙酰乙酸乙酯1.2 mL(9.35 mmol),维持温度0~5 ℃。反应15 min 后,不经处理直接将混合液加入硅胶柱中(PE∶EA=2∶1,Rf=0.3),得黄色5a 固体1g,两步总产率39%。1H NMR(400 MHz,CDCl3)δ:7.66(s,2H,arom),3.39(s,2H,CH2),2.9(m,2H,CH(CH3)2),2.34(s,3H,CH3),2.18(s,3H,CH3),1.21(d,J=6.8Hz,12H,CH(CH3)2).MS(ESI,m/z):317.2[M+H]+。
1.2.1.9 3’⁃甲基⁃5’⁃叔丁基⁃4’⁃乙酰氧基依达拉奉(5b)的合成
合成方法同4a 和5a。3b 0.5 g(2.25 mmol),浓盐酸6 mL,醋酸5 mL,亚硝酸钠0.24 g(3.5 mmol),水0.6 mL,氯化亚锡二水合物1.5 g(6.75 mmol),乙酰乙酸乙酯0.35 mL(2.5 mmol)。经反应并处理后得到淡黄白色固体5b 0.24 g,Rf=0.3(PE∶EA=3∶2),两步产率45%。1H NMR(400 MHz,CDCl3)δ:7.76(d,J=2.8 Hz,2H,arom),7.62(d,J=2.8 Hz,2H,arom),3.41(s,2H,CH2),2.34(s,3H,CH3),2.19(s,3H,CH3),2.14(s,3H,CH3),1.35(s,9H,C(CH3)3).MS(ESI,m/z):325.2[M+Na]+。
1.2.1.10 2’,6’⁃二异丙基⁃4’⁃羟基依达拉奉(6a)的合成
在耐压瓶中依次加入5a 0.4 g(1.26 mmol),2 mol/L氢氧化钠2.5 mL,乙醇7.5 mL,氩气置换后70 ℃反应过夜。反应停止后,3 mol/L 盐酸调节pH=3,乙酸乙酯萃取3次,饱和食盐水洗涤2次,无水硫酸钠充分干燥,旋干得黄色固体产物0.3 g,产率86.7%。1H NMR(400 MHz,CDCl3)δ:7.46(s,2H,arom),4.81(s,1H,OH),3.4(s,2H,CH2),3.16(m,2H,CH(CH3)2),2.19(s,3H,CH3),1.29(d,J=6.8 Hz,12H,CH(CH3)2).MS(ESI,m/z):297.2[M+Na]+。
1.2.1.11 3’⁃甲基⁃5’⁃叔丁基⁃4’⁃羟基依达拉奉(6b)的合成
合成方法同6a。5b 0.24 g(0.8 mmol),2 mol/L氢氧化钠1.6 mL,乙醇4.8 mL。反应处理后得到淡黄白色固体6b 0.1 g,Rf=0.25(PE∶EA=3∶2),产率50%。1H NMR(400 MHz,CDCl3)δ:7.52(d,J=2.8 Hz,2H,arom),7.41(d,J=2.8 Hz,2H,arom),4.74(s,1H,OH),3.4(s,2H,CH2),2.27(s,3H,CH3),2.19(s,3H,CH3),1.42(s,9H,C(CH3)3).MS(ESI,m/z):261.2[M⁃H]-。
1.2.2 生物活性测试
1.2.2.1 DPPH自由基清除实验
DPPH浓度为1.2 mmol/L,样品液配制成各个浓度梯度:0、0.10、0.15、0.20、0.30、0.60、1.20 mmol/L。在一块96 孔板中加入DPPH 溶液500 μL,各个浓度的样品液500 μL,每个浓度的样品溶液设3个平行孔。室温避光孵育30 min,使用酶标仪测定各孔在517 nm 的吸光度。0 mmol/L 样品液的吸光度为A0,其余浓度的吸光度为A,按公式:清除率=(A0-A)/A0×100%计算样品各个浓度的清除率。使用Origin 8.5.1制作样品的清除率曲线,拟合方程Logis⁃tic方程,并得到样品的EC50值。
1.2.2.2 短暂性脑缺血模型
实验方案参照Zhou 等[11]的短暂性脑缺血模型。实验分假手术组、手术组、依达拉奉对照组、化合物6a、6b治疗组,每组动物数15。简而言之,水合氯醛(350 mg/kg,ip)麻醉SD 大鼠,通过颈外动脉残端将4/0 外科尼龙单丝带圆形尖端引入左颈内动脉,在颈动脉分叉处前进20~21 mm,直到感觉到轻微的阻力。此时,腔内细丝阻塞了大脑中动脉、颈内动脉、大脑前动脉和大脑后动脉的所有血源。在整个过程中,体温保持在(37.0±0.5)℃。将栓线留在原位120 min,然后取出再灌注。取出栓线后,立即通过尾静脉给药(药物处方:5% DMSO、18%丙二醇、0.1%亚硫酸氢钠、10% HS⁃15、62%生理盐水)。在假手术动物中,闭塞细丝仅插入颈动脉分叉处上方7 mm处。在MCAO后2 h进行梗死体积测量。快速取出大脑并在-20 ℃下冷冻5 min。制成1~2 mm 冠状切片,切片浸入2% TTC,37 ℃4 h。梗死体积表示为梗死半球中冠状切面的百分比面积。
1.2.2.3 GABA增强作用及直接激动作用实验
采用膜片钳电压钳全细胞记录模式,开始记录之前,小鼠皮层神经元至少观察5 min,保证细胞处于较为稳定状态。灌流液中加入Na+通道阻断剂(TTX,1 μmol/L)、AMPAR 受体阻断剂(CNQX,10 μmol/L)和NMDA 受体阻断剂(APV,50 mmol/L),钳制细胞在-70 mV 记录自发性抑制性突触后电流,根据实验设计,分别灌流GABA或药物,观察其诱发的抑制性反应。记录过程中持续观察串联阻抗,以防止细胞破膜后的重封接,与本底值差异大于20%者弃用。
1.3 统计学方法
应用Excel 分析软件计算测定结果的标准差,实验数据采用均数±标准差()表示,用t检验分析组间差异。用Grubbs 检验法对测定数据的有效性进行分析,以显著性水平α=0.05作为评价数据有效性的标准。
2 结果
从2,6⁃二异丙基苯酚和2⁃甲基⁃6⁃叔丁基苯酚出发,经6 步反应,成功合成了2 个依达拉奉类似物,见图2。DPPH自由基清除实验表明,依达拉奉、6a、6b 的EC50分别为(0.326±0.120)mmol/L、(0.158±0.200)mmol/L和(0.183±0.150)mmol/L(图2)。6a和6b显示了比依达拉奉更强的自由基清除活性。
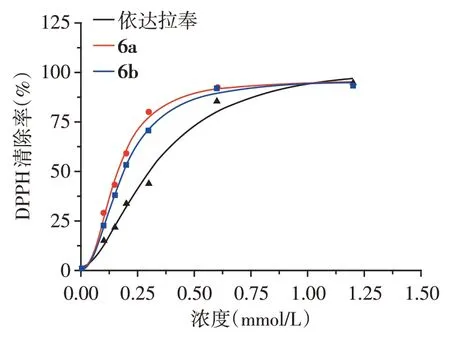
图2 依达拉奉、6a和6b的DPPH自由基清除活性Figure 2 DPPH free radical scavenging activity of edara⁃vone,6a and 6b
体内的脑卒中模型显示,等摩尔量的化合物6a和6b 显示了比对照药物依达拉奉更优的神经保护效果(图3)。手术组的梗死面积百分比是(36.0±3.1)%(n=15),依达拉奉阳性对照组(6 mg/kg)、化合物6a(9.2 mg/kg)、化合物6b(9.44 mg/kg)治疗组的梗死面积百分比分别降为(13.5±1.2)%(n=15)、(10±1.0)%(n=15)、(7.3±0.34)%(n=15)。经t检验,化合物6a和6b的神经保护作用显著优于依达拉奉对照组(P<0.01)。
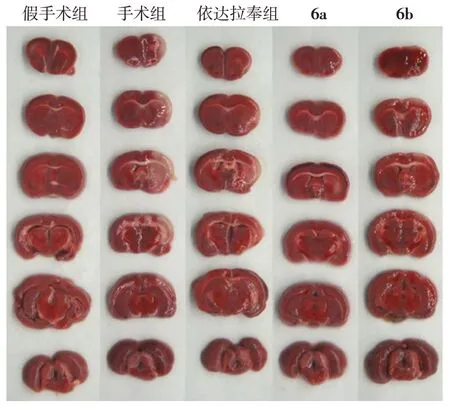
图3 脑切片TTC染色示意图Figure 3 Representative of brain slices by triphenyltetra⁃zolium chloride staining
对神经保护作用较好的化合物6b 进行了GA⁃BA 活性的验证(图4)。化合物6b 在5 mmol/L 时对GABA 没有增强活性,20~40 mmol/L 区间体现显著的GABA 增强活性,且显示剂量依赖性(图4A~C)。6b在高剂量下(500 mmol/L)显示对GABA受体的直接激动活性(图4D)。
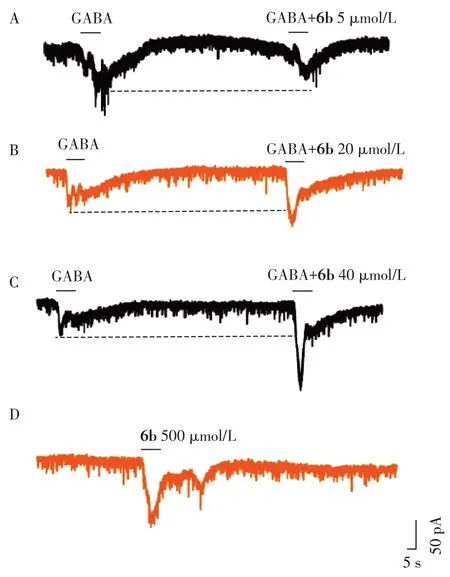
图4 6b的GABA增强活性及直接激动作用Figure 4 GABA⁃enhancing and direct agonistic effect of 6b
3 讨论
脑卒中导致的神经损伤是多因素、多信号通路共同作用的结果,只针对其中一个靶点或者一个信号通路的化合物,往往治疗效果有限,这也许正是脑卒中治疗药物开发难、临床治疗药物缺乏的原因所在[7]。因此,多功能的神经保护试剂必然是今后卒中药物发展的方向[1]。
研究表明,卒中发生时,谷氨酸介导的神经兴奋性毒性在神经损伤中起重要作用[11],而GABA 系统是抑制性神经递质,可拮抗谷氨酸介导的兴奋性损伤,因此能增强GABA活性的物质,具有神经保护作用,例如氯美噻唑曾进入三期临床[7]。近年来,国内批准了先声药业依达拉奉和冰片的复方制剂“先必新®”用于脑卒中的治疗,其中冰片在其中的药理学作用正是GABA 系统的增强剂[12-13]。因此,这些研究成果,为设计多功能的具有GABA 受体增强活性的新型抗脑卒中药物奠定了理论和实践基础。
依达拉奉是一个小分子的抗氧化剂,临床上用作抗脑卒中药物。研究表明,依达拉奉的苯环可被众多的基团修饰而不影响分子的抗氧化性,有的基团取代还能增强分子的抗氧化性[14]。丙泊酚临床上用于麻醉,丙泊酚是GABA 受体的增强剂和激动剂,研究表明,丙泊酚具有神经保护作用,其药理学机制基于其GABA受体激动活性和其自身的抗氧化作用。研究表明丙泊酚的GABA活性对苯环上的取代基团具有较大的耐受性,例如,2⁃甲基⁃6⁃叔丁基的GABA增强活性比丙泊酚还强[15],另外,GABA活性对苯环的4 位取代也具有良好的耐受性。因此,这些研究结果,为设计骨架融合的、具有多功能的新型小分子抗脑卒中药物打下了良好的基础。
在本研究中,利用骨架融合的药物设计原理,概念验证性地合成了2个具有依达拉奉结构和丙泊酚结构或其类似物结构的小分子化合物6a 和6b。由于依达拉奉和丙泊酚类化合物都具有抗氧化活性,因此,在DPPH自由基清除实验中,6a和6b体现了比依达拉奉更强的抗氧化活性。在MCAO 模型中,6a和6b也体现了比依达拉奉更优异的神经保护作用。化合物6b在电生理的实验中,体现了GABA的增强作用和GABAR 的直接激动活性,这些都和丙泊酚及其类似物在低浓度时具有增强作用、在高浓度时具有直接激活作用的现象类似[16]。因此,6b所体现出来的抗氧化活性和GABA活性基本达到了概念验证的目的,具有进一步拓展和深入研究的价值。本研究存在的不足是没有在细胞层面及动物层面对化合物进行自由基清除活性的表征。在今后,计划合成更多的化合物[17],做更深入的药理学研究,例如,在电生理实验中,测定丙泊酚的GABA活性,让化合物的活性比较更为直观。
