实时直接分析-串联质谱法快速测定环境水体中涕灭威及其代谢物
2022-01-26解迎双
解迎双,张 欢,王 娟,王 波
(兰州海关技术中心,甘肃 兰州 730010)
涕灭威是一种氨基甲酸酯类杀虫、杀螨剂,广泛用于多种农作物。涕灭威在喷洒后会迅速氧化生成性质较稳定的涕灭威亚砜,进而氧化生成性质更稳定的涕灭威砜。涕灭威亚砜和涕灭威砜在农业生产中被用作杀虫、杀螨剂[1]。随着人们对健康饮食的需求越来越高,关于食品中涕灭威及其代谢物研究的报道层出不穷,但更多的是关注食品中涕灭威及其代谢物残留所带来的危害,而对于其在环境中造成的危害关注度不够[2-3]。涕灭威及其代谢物在喷洒过程中落入土壤,再通过淋溶作用进入水体,造成对水体环境的污染,对人体的健康存在致癌、致畸、致突变的潜在威胁。世卫组织(WHO)《饮用水水质准则》规定:饮用水中涕灭威(包括涕灭威砜和涕灭威亚砜)的限量为10 μg/L;美国国家环境保护局《饮用水标准和健康指导》规定:饮用水中涕灭威、涕灭威砜、涕灭威亚砜的限值标准分别为3、2和4 μg/L,三者之和不能超过7 μg/L[4]。我国对不同级别地下水中涕灭威的含量进行了限量要求[5]。
目前,涕灭威及其代谢物的检测方法主要有气相色谱法[6]、高效液相色谱法[7]和高效液相色谱-串联质谱法[8-9]。采用气相色谱法检测涕灭威及其代谢物时,因涕灭威热不稳定,造成定量结果不准确,且只能测定涕灭威及其代谢物的总量,不能对单一化合物进行检测。采用高效液相色谱法需要进行柱后衍生,并且需要特殊的衍生装置;柱后衍生会由于衍生不充分而影响方法的灵敏度和重现性,且该方法对涕灭威及其代谢物检测的特异性不强,易产生假阳性。采用高效液相色谱-串联质谱法需要对样品进行预处理,然后进行复杂的色谱分离等过程,步骤繁琐、耗时较长、成本较高[9]。
实时直接分析离子源已广泛用于医疗、环境、食品等领域,但对环境水体中涕灭威及其代谢物的检测鲜有报道[10]。这主要存在2个技术难题:1) 实时直接分析离子源是一种开放性离子源,目标化合物在敞开环境下被离子化后进入质谱仪的量、在TIP头点样位置的不同引起的样品离子化效率的差异,造成了样品间平行性较差。2) 水体样品点样后,若水体样品的溶剂不能完全挥发,在离子化过程中则会消耗过多能量,使目标物质无法得到足够的能量,不能实现充分的离子化,影响检测灵敏度。
基于此,本研究拟通过优化TIP头的点样位置和外置真空泵的真空度,保证准确取样和样品间的平行性;通过优化点样后的溶剂蒸干时间来提高目标物质的离子化效率,从而提高检测灵敏度[11-12]。希望为不同水体环境中相关农药残留的检测提供方法参考。
1 实验部分
1.1 仪器与装置
DART实时直接分析离子源:美国Ion Sense公司产品;三重四极杆质谱仪:美国Waters公司产品;移液器(0.5~10 μL):德国Eppendorf公司产品。
1.2 样品与试剂
涕灭威(100 mg/L)、涕灭威亚砜(100 mg/L)、涕灭威砜(100 mg/L)、乙腈:色谱纯,德国Merck公司产品。
按照《生活饮用水标准检验方法 水样的采集和保存》[13]标准的采样规程,随机采取各10批次兰州市及其周边区县的地下水、二次供水、矿泉水以及城市污水,共计40批次。
1.3 标准储备液与标准工作液的制备
1.3.1标准中间液 分别吸取100 μL 100 mg/L涕灭威、涕灭威亚砜、涕灭威砜于10 mL容量瓶中,用乙腈定容,配制成1 mg/L的3种化合物的单标中间液。
1.3.2标准混合工作液 分别吸取200、100、100 μL 1 mg/L涕灭威、涕灭威亚砜、涕灭威砜于10 mL容量瓶中,用乙腈定容,配制成50、10、10 μg/L标准混合工作液。
1.4 样品处理
地下水、二次供水、矿泉水:直接吸取1 mL样品,过0.22 μm滤膜,准确吸取10 μL样液点至距TIP头1~5 mm处,于35 ℃电热板上氮气吹干后,供实时直接分析-串联质谱仪分析。
城市生活污水:准确吸取5 mL样品于15 mL离心管,以13 000 r/min离心5 min后,取上清液,过0.22 μm滤膜,准确吸取10 μL样液点至距TIP头1~5 mm处,于35 ℃电热板上氮气吹干后,供实时直接分析-串联质谱仪分析。
1.5 实验条件
1.5.1实时直接分析离子源条件 离子化气为氦气,流速2 mL/min;正离子模式检测;12Dip-it Samplers进样模式;点样量10 μL;进样速率0.6 mm/s;离子化温度300 ℃;栅极电压200 V;离子源出口距质谱进样口2~4 cm;外置泵真空度-70 kPa。
1.5.2MS/MS条件 毛细管电压3.2 kV;离子源温度120 ℃;脱溶剂气流速0 L/h;锥孔气流速0 L/h;多反应监测采集模式。优化后的质谱参数列于表1。
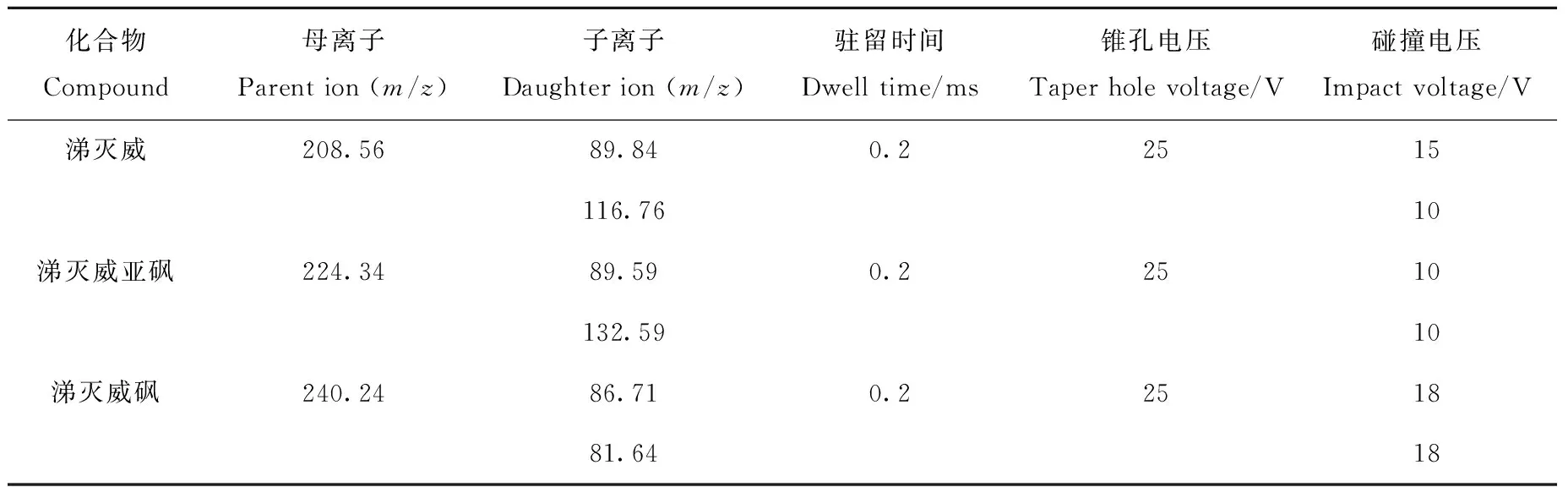
表1 涕灭威及其代谢物的质谱参数Table 1 Mass spectrometric parameters of aldicarb and its metabolites
2 结果与讨论
2.1 质谱条件的优化
2.1.1离子化气体温度的选择 离子化气体温度会影响离子化程度,本实验在250~400 ℃范围内对离子化气体温度进行优化,涕灭威及其代谢物的定量离子峰面积曲线示于图1a。可见,同一浓度的3种目标化合物的峰面积随着离子化气体温度的升高先增加后减小,当达到300 ℃时,目标化合物的峰面积最高。这主要是因为较高的电离温度可以加速样品的热解吸,增加单位时间内进入质谱仪的带电离子数量,从而增强分析物的响应。但是,温度过高会使化合物过早脱附,降低了灵敏度,当离子化气体温度达到350 ℃时,3种目标化合物的峰面积急剧下降。因此,本研究选择300 ℃为最佳的离子化气体温度。
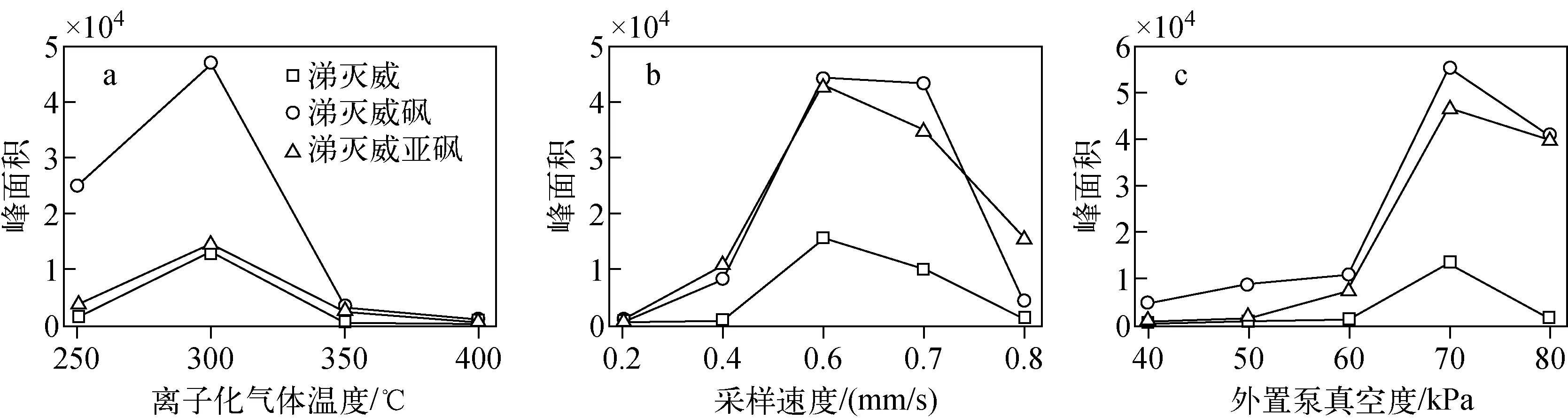
图1 不同气体温度(a)、进样速度(b)、真空度(c)下,涕灭威及其代谢物的定量离子峰强度响应图Fig.1 Quantitative ion peak intensity of aldicarb and its metabolites with different gas temperatures (a), injection rates (b), vacuum degrees (c)
2.1.2进样速度的选择 本研究采样方式为将点有样品的TIP头置于自动采样器的采样架上,当自动采样器匀速经过实时直接分析离子源的离子化区域时,样品被离子化,然后通过陶瓷管取样口进入三重四极杆质谱仪分析。在此过程中,样品的进样速度与样品在离子化区域的通过时间呈反比[12-14],从而对样品的离子化效率产生影响。当进样速度较慢时,样品离子化时间增加,容易造成峰展宽,影响定量准确性。反之,当进样速度过快时,样品在离子化区域停留时间过短,使样品不能完全解吸附,从而降低灵敏度。以3种目标化合物定量离子的峰面积为指标,在0.2~0.8 mm/s范围内对进样速度进行优化,结果示于图1b。可以看出,当进样速度为0.6 mm/s时,涕灭威及其代谢物的定量离子峰面积最大。
2.1.3外置真空泵真空度的选择 实时直接分析离子源与三重四极杆的接口为敞开式,并且以3.0 L/min向质谱仪取样口吹氦气或氮气,因此,三重四极杆的真空度会受到影响,而实时直接分析离子源搭配的外置真空泵可以稳定三重四极杆质谱仪的真空度[15]。样品在稳定的真空度作用下被带入取样口,保证了实验的灵敏度和平行性,但真空度过高会使样品在进入质谱仪的过程中被抽离质谱,降低了检测灵敏度。以3种目标化合物定量离子的峰面积为指标,在-40~-80 kPa范围内对外置真空泵的真空度进行优化,结果示于图1c,当真空度为-70 kPa时,涕灭威及其代谢物的响应值最佳。
2.1.4点样量及点样方式的选择 分别采用同一浓度的混合标准物质,在其他参数不变的情况下对点样量进行优化。本实验对比了5、10、15、20 μL点样量的离子峰面积,示于图2a。可见,点样量为10 μL较5 μL的目标物响应明显增加;当点样量为15 μL和20 μL时,其结果与10 μL时差别不大,可能是由于点样量大于10 μL时,超出了TIP头正面的承载能力,点样后的溶剂会随TIP头扩散至TIP头背面,影响了离子化效率。点样量在10~20 μL范围内均可满足检测要求,但以点样量为10 μL时响应最佳。
以10 μL水体样品点样后,样液晾干时间较长,晾干后样品损失较大,影响检测灵敏度。本实验选择乙腈和水作为溶剂,分别配制100 μL涕灭威及其代谢物的标准混合溶液,结果表明,以乙腈为溶剂的标准混合溶液点样后的晾干速度更快,目标物响应更好。为进一步提高晾干速度,经优化后,本实验选择将水体样品点样后,于45 ℃加热板加热,再以氮气流吹扫进行样品晾干,晾干时间控制在5 min内。结果表明,目标物响应明显增加,可满足国家标准要求的检测灵敏度。
2.1.5点样位置的选择 当加载有样品的TIP头匀速经过离子化区域时,只有当点样位置在离子源和三重四极杆质谱仪同一水平时,样品才可以实现最大程度离子化,且点样位置的不同会影响样品间的平行性。当点样位置出现以下2种情况时,样品不能被有效离子化,会影响检测灵敏度和样品间的平行性:1) 在TIP头最下端点样时,样液易扩散至TIP头末端及背部;2) 当在5 mm以上位置点样时,超出了离子化区域的高度。点样位置在距离TIP头下端1~5 mm之间时,离子化效果最佳,且样品间的平行性最好,示于图2b。
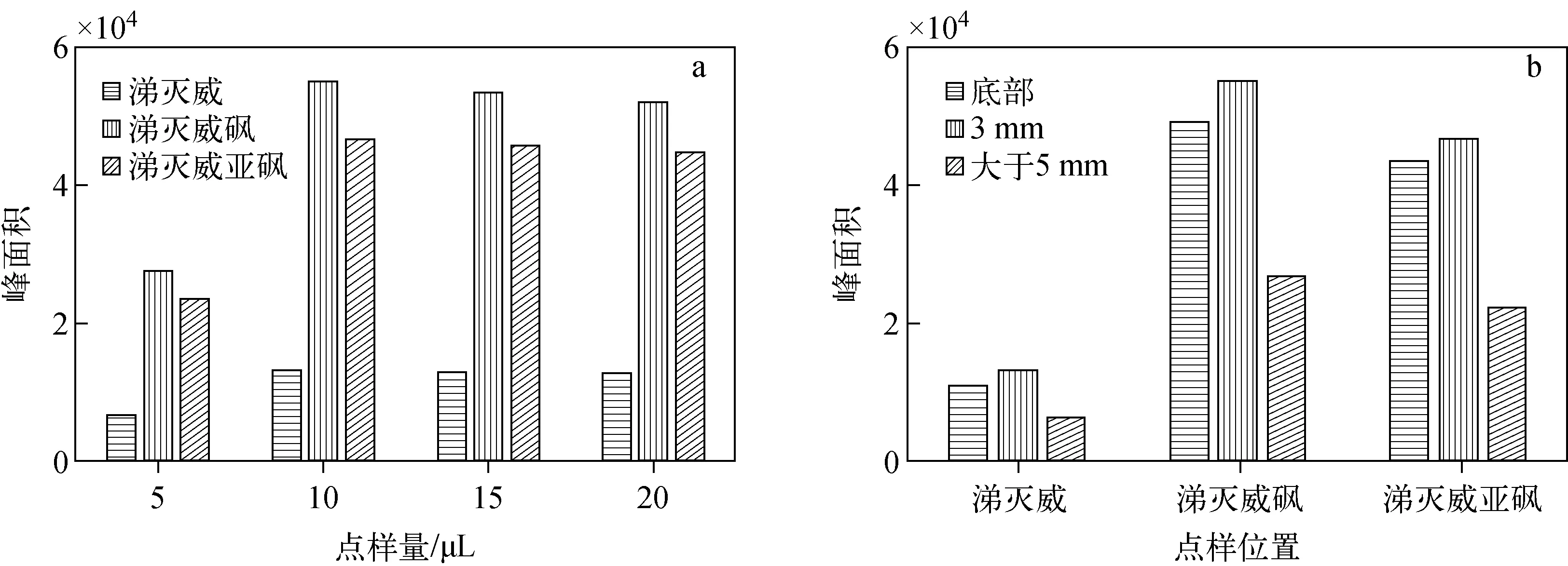
图2 不同点样量(a)和不同点样位置(b)下,涕灭威及其代谢物的定量离子峰强度Fig.2 Quantitative ion peak intensity of aldicarb and its metabolites with different sample sizes (a) and sites (b)
2.1.6TIP头抗污染能力实验 本方法未进行样品前处理,为防止基质复杂的样品对仪器的灵敏度和稳定性造成影响,进行了优化实验:1) 通过对TIP头1次点样后多次进样,考察TIP头上的样品残留所造成的污染,实验表明,重复4次进样后依然有样品残留,将TIP头经乙腈浸泡、超声后再次进样后未发现样品残留,因此,本研究所用的TIP头经1次点样后,采用乙腈浸泡30 min,再超声10 min后可重复利用;2) 通过每隔5个标准溶液穿插2个空白TIP头进样,不会对仪器造成残留影响。
2.2 方法学考察
2.2.1方法检出限、线性范围和精密度 本实验将配制的不同浓度的目标物混合标准溶液添加到4种类型的水体样品中,同一样品平行测定10次,将平均值作为最终数据,以3倍信噪比计算检出限,10倍信噪比计算定量限,其结果列于表2,示于图3。在优化的仪器条件下测定配制的系列标准溶液,以目标化合物的质量浓度(x,μg/L)对其峰面积(y)进行线性回归,各化合物的线性范围、相关系数及线性回归方程列于表2。3种目标物的相关系数均大于0.98。在10 μg/L加标水平下进行6次平行实验,相对标准偏差<10%,能够满足检测要求。

表2 涕灭威及其代谢物的线性范围、检出限、定量限、精密度Table 2 Linear range, detection limit, quantification limit and precision of aldicarb and its metabolites
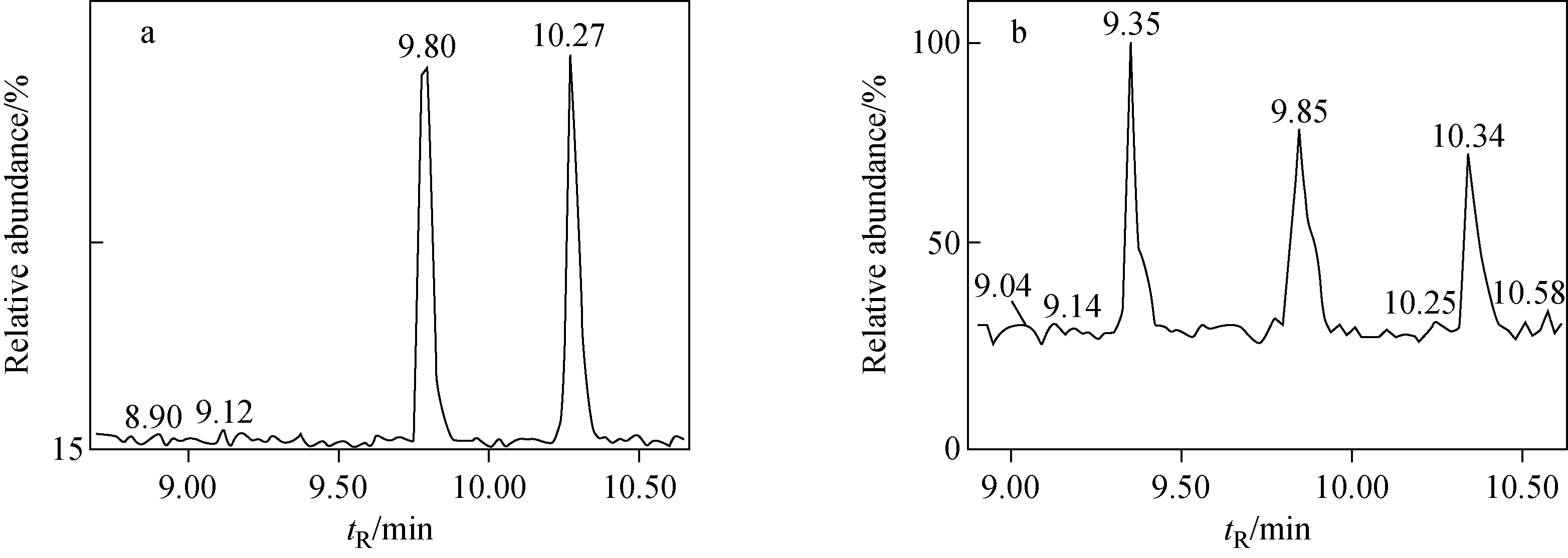
图3 矿泉水(a)、城市污水(b)中涕灭威及其代谢物检出限混合标准溶液平行2针谱图Fig.3 Chromatograms of aldicarb and its metabolites in mineral water (a) and municipal wastewater (b) by two-needle parallel
2.2.2加标回收率 鉴于DART原位电离的特点,本方法对回收率的影响因素仅体现在离子化效率。为验证方法的准确性,本研究选取了3个不同的样品浓度值,按照1.3~1.5节方法对同一样品平行测试6次后,计算得到的回收率,列于表2。
2.2.3抗干扰能力实验和仪器稳定性分析
本方法在检测过程中未对样品进行色谱分离,容易产生假阳性。选择三重四极杆质谱多反应监测(MRM)模式,采用1个母离子对应2个子离子的方式对所测化合物进行二次分离。以本研究的阴性水体样品为溶剂配制的检出限基质标准溶液、10 μg/L基质标准溶液、100 μg/L基质标准溶液分别重复进样12次后,计算涕灭威及其代谢物的2个定性离子丰度比的平均值。以离子丰度值结合国际对离子丰度比最大偏差允许值的范围作为该化合物的抗干扰指标,列于表3。结果表明,涕灭威、涕灭威亚砜、涕灭威砜的定性离子对丰度比平均值分别为2.06、3.23、2.57,离子对丰度比K<10,允许最大偏差为±50。

表3 定性确证时,相对离子丰度的最大偏差Table 3 Maximum deviation of relative ion abundance for qualitative confirmation
2.3 基于实时直接分析离子源下涕灭威及其代谢物的裂解规律分析
涕灭威、涕灭威砜、涕灭威亚砜均属于氨基甲酸酯类化合物,含有R—COO—R结构,当样品基质为水体时,采用实时直接分析离子源进行检测易发生加合反应,形成[M+NH4]+[15]。现对3个化合物的碎裂机理进行推测。
2.3.1涕灭威的裂解规律 涕灭威的分子式是C7H14N2O2S,相对分子质量为190.29。将准分子离子m/z208.5 [M+NH4]+裂解后,得到的主要碎片离子为m/z143.48、116.46、74.56和86.75,示于图4a。其中,m/z86.75可能为含硫巫类氨基甲酸酯类农药的特征峰[CHN2HCOO]+;m/z74.56和m/z116.46可能为涕灭威经过C-N键断裂后生成的[CH3NHCOO]+和[CH3SC-CH3CH3CNH]+;m/z143.48可能为涕灭威结构中C—S键断裂后丢掉[CH3-S]+形成的。
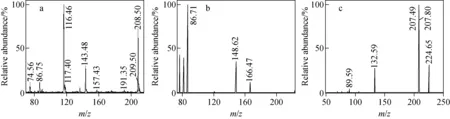
图4 涕灭威(a)、涕灭威砜(b)、涕灭威亚砜(c)的二级质谱图Fig.4 MS/MS spectra of aldicarb (a), aldicarb sulfone (b), aldicarb sulfoxide (c)
2.3.2涕灭威砜的裂解规律 涕灭威砜的分子式是C7H14N2O4S,相对分子质量为222.26。将准分子离子m/z240.24 [M+NH4]+裂解后,得到主要碎片离子m/z166.41、148.62、86.71、81.64、76.70,示于图4b。其中,m/z166.41可能为涕灭威砜结构中C—S键发生断裂后形成的碎片加合Na+离子峰[CH3NHCOONCHCCH3CH2Na]+;m/z148.62可能为涕灭威砜结构中丢掉[CH3NHCOO]+后形成的碎片;m/z81.64可能为涕灭威砜结构中C—S键发生断裂后形成的碎片加合2个H+离子峰[CH3OOSH]+;m/z76.70可能为涕灭威砜结构中[CH3NHCOO]+加合2个H+离子之后形成的碎片。
2.3.3涕灭威亚砜的裂解规律 涕灭威亚砜的分子式是C7H14N2O3S,相对分子质量为206.26。将准分子离子峰m/z224.34 [M+NH4]+裂解后,得到的主要碎片离子为m/z207.49、132.59、89.59,示于图4c。m/z207.49可能是[M+H]+峰;m/z132.59可能为该化合物丢掉[CH3NHCOO]+后形成的碎片;m/z89.59可能是碎片m/z132.5产生中性丢失后,丢掉1个CO2形成的碎片。
综上所述,在实时直接分析离子源条件下,涕灭威、涕灭威砜、涕灭威亚砜的常见裂解规律为:一级质谱条件下易加合NH4+离子形成[M+NH4]+峰;在断裂过程中易发生C—S键断裂、易丢失[CH3NHCOO]+、易形成硫巫类化合物的特征峰m/z86.50。在未来工作中,将结合MassworksTM等软件对涕灭威、涕灭威砜、涕灭威亚砜在实时直接分析离子源条件下的断裂规律进行验证。
2.4 对比DART-MS/MS和LC-MS/MS方法检测实际样品
2.4.1方法对比结果 从表4可以看出,国标GB/T 23214方法中水体样品预处理繁琐[16],需要11种试剂,单个样品的试剂总消耗量为71 mL,且分析耗时较长[17]。本方法简化了检测流程、缩短了检测时间、实现了样品的高通量检测,同时节约试剂,降低成本,减少对环境的二次污染,极少产生加和离子,极少形成源外电离,可以不进行色谱分离即可根据分子质量对混标样品中的不同化合物进行定性。将实时直接分析离子源与MS/MS串联后,可发挥多反应监测功能,通过碎片离子的不同,实现化合物的二次分离和准确定性,极大地提高了该技术的检测灵敏度[18-22]。单个样品的分析时间只有几秒钟,实现了水体样品的原位高通量检测,且本方法的检出限可达到国家标准要求。

表4 国标方法与实时直接分析离子源-串联质谱法的对比Table 4 Comparison of national standard method and real-time direct ion source tandem mass spectrometry
2.4.2实际样品测定结果 为了进一步验证本方法的准确性,随机抽取了兰州市部分地下水样品,采用国标方法和本方法进行检测,3种目标物质均未检出。
3 结论
本研究采用实时直接分析法快速检测环境水体中涕灭威及其代谢物。通过优化点样位置、点样方法和离子化效率等参数,克服了DART离子源取样不均、平行性较差的缺点,可实现定量分析。本方法基本无需样品前处理,不消耗有机溶剂,操作简单、省时,能同时检测环境水体中涕灭威及其代谢物,并可实现高通量检测,为快速准确检测环境水体中的农药残留提供了手段和思路。在未来工作中,将结合MassworksTM等软件对涕灭威、涕灭威砜、涕灭威亚砜在实时直接分析离子源条件下的断裂规律进行验证。
