微囊藻毒素-LR 致生殖细胞凋亡的分子机制研究进展
2020-10-22杜星德张慧珍
杜星德, 张慧珍
(郑州大学公共卫生学院, 河南 郑州450001)

微囊藻毒素 (microcystins, MCs) 是一类分布最广泛的蓝藻毒素, 主要存在于富营养化水体中,具有环状七肽结构, 可溶于水, 易溶于有机溶剂,化学性质稳定[1]。 MCs 可在水体中存在数月至数年,现有的饮用水处理方法不能有效去除水中的MCs, 对人类的健康造成了极大威胁。 目前已经鉴定出超过200 种MCs 的同分异构体, 其中微囊藻毒素-LR (microcystin-LR,MC-LR) 是毒性最强, 也是含量最多的一种亚型[2]。 国际癌症研究机构(International Agency for Research on Cancer, IARC) 早在2010 年便已将MC-LR 列为“人类可疑致癌物”[3]。 为了减少MC-LR对人体健康带来的风险, 世界卫生组织规定, 饮用水中的MC-LR 的浓度不得超过1 μg/L[4]。 MCs 具有多器官毒性, 肝脏是其首要靶器官, 性腺是第二靶器官[5]。 MC-LR 能够在哺乳动物、 两栖动物、 鱼类、 鸟类等多种动物的生殖系统中蓄积并产生明显的生殖毒性[5]。 MC-LR 作为肿瘤的启动剂和细胞凋亡的诱导剂, 可通过细胞膜上的有机阴离子转运多肽进入细胞内发挥毒性作用[6]。 MC-LR 诱导生殖细胞凋亡是其发挥毒性的重要过程之一, 细胞凋亡是一种受基因控制的程序性死亡方式, 此过程涉及一系列基因的激活、表达以及调控等。 进入细胞内的MC-LR 主要通过抑制蛋白磷酸酶活性、 氧化应激损伤以及通过对相关基因进行表观遗传修饰来诱导生殖细胞凋亡。本文就MC-LR 诱导生殖细胞凋亡的分子机制研究进展做一综述。
1 抑制蛋白磷酸酶活性
蛋白磷酸酶1 和2A (protein phosphatase 1/2A, PP1/2A) 同属于蛋白磷酸酶家族, MC-LR对PP2A 的亲和力和抑制能力均比PP1 高, 因此研究PP2A 在MC-LR 诱导的毒性中的作用更具代表意义[7]。 PP2A 是一种在细胞内执行丝氨酸/苏氨酸蛋白的脱磷酸化功能的蛋白磷酸酶, 对调控细胞周期、 维持细胞骨架和细胞内的信号转导起关键作用[8]。 MC-LR 作为一种特异性的PP2A 抑制剂, 可以通过与PP2A 催化亚基的活性位点结合来抑制PP2A 活性, 导致细胞内磷酸化和脱磷酸化水平失衡进而产生毒性作用[1]。 MC-LR 还可以通过抑制PP2A 的活性使下游信号通路中的关键蛋白发生磷酸化, 从而激活凋亡相关通路, 诱导生殖细胞凋亡, 产生生殖毒性。 研究显示, 实验小鼠暴露于MC-LR 后, 在睾丸中发现p53 和Bcl-2 的磷酸化水平显著增加以及细胞凋亡的发生, 表明蛋白质的磷酸化可能参与了MC-LR 诱导的细胞凋亡[9]。 Ca2+/钙调蛋白依赖性蛋白激酶Ⅱ(CaMKⅡ) 信号通路和丝裂原激活蛋白激酶(MAPK)信号通路是调控细胞凋亡的两条重要通路[7]。 在MC-LR 暴露下, CaMKⅡ和MAPK 信号通路功能缺失细胞的凋亡率显著降低[10-11]。 这些研究表明,MC-LR 在生殖细胞内通过抑制PP2A 活性, 使CaMKⅡ和MAPK 等凋亡相关通路中的关键蛋白发生磷酸化而激活, 最终诱导生殖细胞的凋亡。 然而, 细胞内还存在着诸多抗凋亡通路(如JAK/STAT、 NF-κB、 PI3K/Akt 等) 同样可通过磷酸化激活[12-14], 这些抗凋亡通路在MCs 的生殖毒性研究领域同样具有重要意义, 此类促凋亡与抗凋亡通路之间的联系将是我们下一步研究的重点, 且MC-LR 暴露于生殖细胞后, 凋亡相关基因的表达机制尚未明确, 此部分有待深入探索。
2 氧化应激损伤
氧化应激是指机体在遭受有害刺激时, 体内活性氧自由基(reactive oxide species, ROS) 等高活性分子过量产生, 氧化程度超出机体对氧化物的清除能力, 氧化系统和抗氧化系统失衡引起的应激反应。 氧化应激被认为是MC-LR 发挥毒性的始发因素[15]。 长期的氧化应激可以造成细胞中蛋白质结构损害、 内质网和线粒体等细胞器损伤,进而诱导细胞凋亡的发生。 而内质网与线粒体是细胞凋亡信号传导途径中具有重要作用的细胞器[16]。 性腺富含不饱和脂质, 易受氧化应激损伤[5]。
2.1 内质网途径 细胞在受到MC-LR 的刺激时,会产生高水平的ROS。 当机体内ROS 的水平超出了抗氧化能力时, 便会加重氧化应激反应, 并诱导内质网的结构和功能损伤, 最终导致错误折叠蛋白以及未折叠蛋白的大量堆积, 进而发生内质网应激(endoplasmic reticulum stress, ERS)[17-18]。若ERS 强度过大、 时间过长, 便会诱导细胞凋亡的发生。 研究发现, MC-LR 可以诱导C57BL/6 小鼠卵巢颗粒细胞(KK-1) 产生大量的ROS, 并诱导分子伴侣葡萄糖调节蛋白78 (GRP78)、 X 盒结合蛋白1 (XBP1) 和C/EBP 同源蛋白(CHOP)等ERS 相关蛋白的表达升高, 而在使用抗氧化剂N-乙酰半胱氨酸(N-Acetyl-L-cysteine, NAC)进行干预后, 明显减弱了ERS 相关蛋白的表达水平, 这说明氧化应激介导了MC -LR 诱导的ERS[19]。 为了修复由ERS 所引起的细胞损伤, 恢复细胞功能, 未折叠蛋白反应 (unfolded protein response, UPR) 便会触发。 UPR 的信号转导由三个跨膜蛋白调控, 分别是蛋白激酶R 样内质网激酶(PERK)、 需肌醇酶1 (IRE1) 和转录激活因子6 (ATF6)[16]。 在正常生理条件下, 这些跨膜蛋白与内质网腔中的GRP78 相结合, 使ATF6、IRE1 和PERK 信号通路处于静息状态。 当ERS 发生时, 未折叠蛋白在内质网中积累, 此时GRP78从这些复合物中释放出来, 与新的未折叠蛋白结合, 从而使ATF6、 IRE1 和PERK 三条信号通路被激活[20]。 这些三条通路的激活顺序是不同的,GRP78 解离激活PERK 信号通路几乎是与ERS 同时发生, 随后ATF6 信号通路的激活, 最后是IRE1 信号通路[21]。 PERK 与GRP78 分离后, 发生自身磷酸化而活化, 活化的PERK 会引起真核起始因子2 的α-亚基(eIF2α) 的磷酸化, 磷酸化的eIF2α 通过激活转录激活因子4 (ATF4) 来增高CHOP 表达, 从而引起细胞凋亡[22-23]。 ATF6 是参与UPR 的关键蛋白, ERS 时, ATF6 被切割后成为具有转录因子活性的蛋白, 并从高尔基体膜转位至细胞核中, 进而激活下游的XBP1 和CHOP, 诱导 细胞发生凋亡[24]。 最后, IRE1 与GRP78 迅速解离, 通过自身磷酸化而活化后, 会引起XBP1 mRNA 的转录水平增加, 然后IRE1 的核糖核酸内切酶结构域(RNase) 会将XBP1 特定位点上的26 个核苷酸剪切掉, 使之变成剪切型XBP1 (XBP1s)[25]。 XBP1s 是UPR 中的转录激活因子, 具有促进内质网分子伴侣和UPR 靶蛋白等UPR 效应基因的转录的功能, 在蛋白质折叠、 加工和运输, 以及降解错误折叠的蛋白质过程中发挥重要作用。 如果内质网中的未折叠蛋白或错误折叠蛋白未能被UPR 缓解, IRE1 的持续激活会导致JNK 信号通路被激活, 最终诱导细胞凋亡的结局[26]。 研究发现, MC-LR 可以通过ERS 诱导中国仓鼠卵巢(CHO) 细胞发生凋亡, 使ERS 标志性蛋白GRP78、 ATF6、 PERK、 IRE1、 CHOP 和自噬蛋白Beclin1、 LC3Ⅱ的表达水平随着MC-LR 染毒浓度升高而增加, 而在对细胞预先使用ERS 抑制剂(4-PBA) 处理后, 细胞凋亡率出现明显下降,这说明ERS 介导了MC-LR 诱导的细胞凋亡[27]。
内质网不仅是蛋白质合成和加工的场所, 同时也是细胞内Ca2+存储和钙信号转导的主要部位,细胞内Ca2+常作为第二信使参与细胞凋亡过程[28],研究显示, MC-LR 可以诱导细胞内Ca2+升高, 导致Ca2+稳态失衡, Ca2+水平的上升又会激活CaMKⅡ信号通路, 此机制似乎在MC-LR 诱导的细胞凋亡中起着关键作用[27,29]。 尽管诸多研究认为内质网作为细胞内的“钙库” 与Ca2+稳态密切相关, 然而MC-LR 诱导的Ca2+稳态失衡是归咎于ERS 引起的内质网结构功能破坏, 还是由于内质网膜上与Ca2+摄入和释放相关的通道受体功能异常所致? 亦或是受其他因素调控? 此方面的研究仍需要大量的实验数据来支持。
2.2 线粒体途径 线粒体介导的凋亡通路主要是由MC-LR 诱导细胞内产生的过量ROS 作用于线粒体外膜, 导致线粒体膜通透性转换孔 (mitochondrial permeablity transition pore, mPTP) 开放,引起线粒体膜电位降低, 破坏线粒体结构等, 从而将线粒体内的凋亡诱导因子(AIF) 和细胞色素C 等释放到线粒体外, 促使下游Caspase 家族蛋白活化或者直接作用于相应底物, 启动细胞凋亡程序[30-33]。 线粒体外膜通透性受Bcl-2 家族成员调控。 Bcl-2 家族成员根据其特性通常被分为抗凋亡成员如Bcl-2、 Bcl-xL 等, 以及促细胞凋亡成员如Bax、 Bak、 Bid 和Puma 等[34]。 Bcl-2 可以通过与Bax 竞争性的同mPTP 结合, 抑制mPTP 的开放,进而调控细胞凋亡的过程。 正常生理条件下,mPTP 通过周期性开放维持线粒体的正常功能, 而ROS 的持续蓄积可导致mPTP 的过度开放从而诱导细胞凋亡的发生[32,35]。 研究显示, MC-LR 可以诱导SD 大鼠生精细胞和支持细胞中ROS 生成,激活Bcl-2 家族, 降低线粒体膜电位, 使细胞色素C 释放进入细胞质, 从而激活Caspase 家族的相关成员, 诱导细胞凋亡的发生; 而抗氧化剂NAC或白藜芦醇预处理细胞后, ROS 的生成减少, 抗凋亡蛋白表达升高, 促凋亡蛋白表达下降, 线粒体损伤得到缓解, 细胞凋亡率出现明显下降, 这说明氧化应激在MC-LR 通过线粒体途径致细胞凋亡过程中具有重要作用[36-38]。 mPTP 定位于线粒体内外膜之间, 是一种由多种跨膜蛋白(ANT、VDAC、 PiC 和Cyp D 等) 组成的蛋白复合体, 它们直接或间接参与mPTP 的形成和开放[39]。 mPTP参与了细胞死亡过程中线粒体成分的释放, 在细胞凋亡的进程中扮演着重要角色。 环孢菌素A(ciclosporin A, CsA) 作为保护mPTP 正常功能的有效药物, 可以抑制细胞色素C 从线粒体释放到细胞质。 使用CsA 预处理大鼠睾丸支持-生精共培养细胞后, MC-LR 诱导的线粒体损伤得到显著改善, 有效阻止了细胞色素C 自线粒体的释放,线粒体膜电位得到恢复, 修复了线粒体功能损伤,促凋亡相关蛋白的表达水平降低, 细胞的凋亡率下降, 提示mPTP 在MC-LR 通过线粒体途径致细胞凋亡过程中具有重要作用[40]。
线粒体呼吸链是内源性ROS 的最主要来源,因此线粒体更易受到氧化损伤[41]。 现阶段研究证实了MC-LR 可以在细胞中诱导线粒体损伤, 破坏线粒体的膜结构[42-43], 并通过线粒体途径诱导细胞凋亡的发生, 进而发挥生殖毒性, 这体现了线粒体在细胞的信号转导过程的重要作用。 同时,线粒体作为细胞能量供应单元, 在生精过程(即精子发生过程, 指精原细胞通过逐级分化、 分裂形成精子的过程) 中起到能量供应的作用[44]。 因此我们推测, 生殖细胞的凋亡除了受线粒体信号转导过程影响外, 还可能因线粒体受损导致的细胞能量供应不足而引起。 此外, 线粒体自噬作为清除受损线粒体, 维持细胞稳态的主要机制, 引起了人们的关注。 那么MC-LR 能否诱导线粒体自噬的发生? 线粒体自噬与细胞凋亡又存在怎样的关系? 这一系列的问题还需解答。
3 表观遗传修饰
表观遗传修饰是调节转录的一种常见机制,对调节细胞周期、 维持细胞稳态和调控程序性细胞死亡具有重要作用[45]。 越来越多的证据表明,表观遗传调控凋亡相关基因的表达在细胞凋亡过程中起着重要作用[46]。 在细胞凋亡过程中, p53是多种细胞应激信号的中心传感器, 当p53 活性增加时, 可以引起细胞周期阻滞, 并通过多种途径导致细胞凋亡。 研究表明, MC-LR 可以增加大鼠睾丸支持细胞中促凋亡相关蛋白p53 的表达,通过线粒体Caspase 依赖途径增加Caspase-3 的活性, 诱导细胞发生凋亡[36,40]。 而包括组蛋白的甲基化和乙酰化在内的表观遗传修饰可以调控p53的表达, 进而诱导细胞凋亡[47-49], 提示表观遗传修饰调控凋亡相关基因的表达可能是MC-LR 诱导细胞凋亡的一种新机制。 组蛋白甲基化与凋亡相关基因启动子区域的转录激活密切相关。 组蛋白第三亚基四号赖氨酸的三甲基化(H3K4me3) 通常被认为是p53 的活化信号, 在细胞遗传毒性应激响应中, H3K4me3 可通过与TAF3/TFIID 的相互作用增强p53 及其下游调控基因的表达[47]。 研究发现H3K4me3 在MC-LR 诱导的生殖毒性中起重要作用。 MC-LR 诱导p53 表达水平的升高可能是由于H3K4 在生殖细胞中的甲基化所致[38]。 组蛋白乙酰化是表观遗传修饰的另一种重要作用方式, 与调控基因活性关系密切, 对真核生物基因的表达和沉默具有重要作用。 乙酰化修饰在细胞周期、 细胞分化、 细胞增殖以及凋亡等生物学过程中发挥着关键作用。 组蛋白乙酰化、 去乙酰化之间动态平衡的打破会引起基因表达调控紊乱,造成细胞周期、 基因转录的异常, 进而引起细胞凋亡的发生[46]。 研究发现组蛋白的乙酰化水平升高, 能促进抑癌基因p53 活化和表达, 下调Bcl-xL以及上调Bax 的表达水平, 促进细胞凋亡的发生[48]。 在体内和体外的研究也已证实, MCLR 可引起SD 大鼠睾丸组织和睾丸支持-生精共培养细胞组蛋白乙酰化酶活性改变, 影响组蛋白乙酰化水平, 进而通过激活线粒体Caspase 通路、干扰细胞周期诱导SD 大鼠睾丸组织和睾丸支持—生精共培养细胞凋亡的发生[50]。
表观遗传修饰是基因表达调控的重要组成部分, 主要包括DNA 甲基化、 组蛋白修饰和非编码RNA 等[45], 虽然现阶段研究主要发现组蛋白修饰参与了MC-LR 诱导生殖细胞凋亡的过程, 但最新研究显示, MC-LR 可以诱导肝细胞DNA 甲基化和miRNAs 的改变[49,51]。 还有研究发现, MC-LR慢性暴露后, 小鼠的睾丸内多种非编码RNA(miRNAs、 lncRNAs、 circRNAs 和piRNAs 等) 出现差异表达, 但这些非编码RNA 异常表达的原因尚未进行探寻[52]。 总之, 这些数据提示包括DNA甲基化和非编码RNA 改变在内的表观遗传学机制可能也参与了MC-LR 诱导的生殖毒性。
综上所述, MC-LR 主要是通过抑制蛋白磷酸酶的活性、 氧化应激损伤以及表观遗传修饰诱导生殖细胞凋亡, 见图1。 目前已有大量学者对MC-LR 诱导生殖毒性进行了详尽的实验研究, 然而仍存在诸多值得探索的地方, 已在上文中讨论。同时, 除了细胞凋亡外, 还存在其他多种受调控的程序性细胞死亡方式在机体的生命活动中发挥作用, 如坏死性凋亡 (necroptosis)、 细胞焦亡(pyroptosis) 以 及 铁 死 亡 (ferroptosis) 等[53],MC-LR是否通过其他受调控的细胞死亡方式发挥其毒性作用仍有待研究。 总之, MC-LR 诱导生殖细胞凋亡的机制研究极为复杂, 并可能存在多条通路互相影响、 互相干扰的情况, 因此在研究过程中要对通路中的关键分子进行深入的挖掘和探索, 唯有如此, 才能全面、 深入地阐述MC-LR 诱导生殖细胞凋亡的机制。
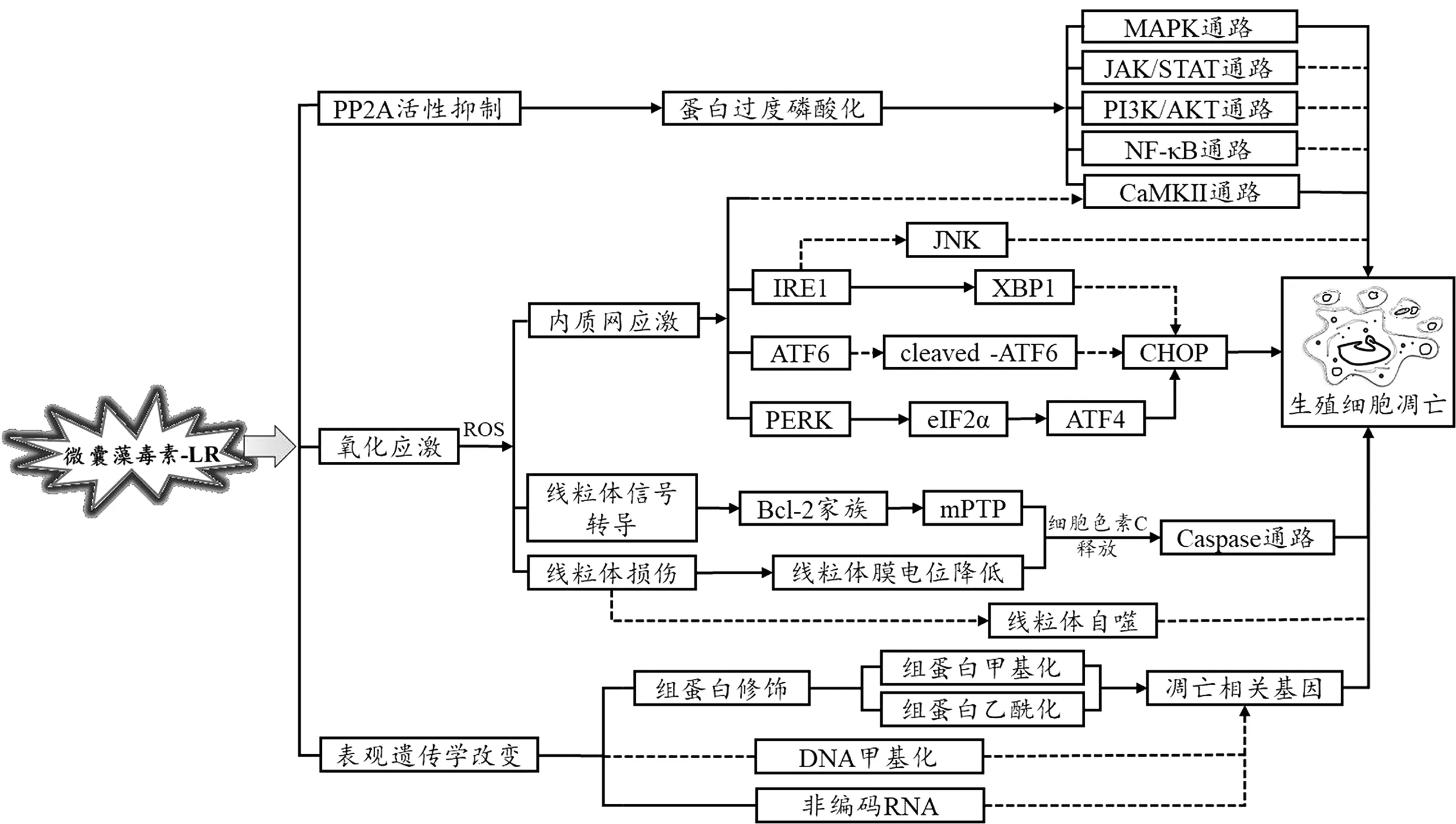
图1 微囊藻毒素-LR (MC-LR) 致生殖细胞凋亡过程中可能的机制图(实线部分表示已被证实的机制,虚线部分表示可能存在但尚未得到验证的机制)
