猪伪狂犬病病毒流行株gE/gI双基因缺失突变株的构建及纯化
2020-03-12郑慧华田润博徐朋丽韩昊莹张鸿鑫崔建涛陈红英河南农业大学牧医工程学院河南郑州450002
张 宇,郑慧华,田润博,赵 宇,徐朋丽,韩昊莹,张鸿鑫,崔建涛,陈红英 (河南农业大学 牧医工程学院,河南 郑州450002)
猪伪狂犬病(porcine pseudorabies,PR),其病原是猪伪狂犬病病毒(porcine pseudorabies virus,PRV),主要症状为发热、奇痒、脑脊髓炎、神经及繁殖系统障碍,是一种急性传染病[1-2]。目前尚无研发出可成功治疗PR 的药物,只能在临床上采用接种弱毒疫苗来对PR 进行防控[3-4]。我国于上世纪70年代从匈牙利引进了PRV Bartha-K61株[5-6],一直以来国内猪场普遍使用该疫苗,使PR 在我国得到了有效的控制。
但2011年后,我国多个省份中已免疫过PR 疫苗的猪群中出现了新一轮PR 疫情的大流行[7-8]。许多研究者也陆续从发生PR 新疫情、免疫过PR 疫苗猪场的死亡仔猪和流产胎儿中分离到了PRV 流行株,研究结果也证实新流行株的抗原性已经发生变异,且对仔猪的致病性有所增强,现有的经典PRV Bartha-K61弱毒疫苗株不能完全保护目前流行株的感染[3,9-10]。这一轮以PRV 流行株引发的PR 疫情使我国的养猪业面临巨大的风险,因此,急待开发一种针对当前流行株的疫苗[11-12]。
本研究以PRV NY 株为亲本毒株,参照PRV经典疫苗株Bartha-K61株的基因缺失部位,釆用同源重组,同时结合蚀斑筛选纯化技术,成功构建了1株针对目前PRV 流行株的gE/gI双基因缺失病毒,以期将其用于防控当前PRV 流行株引起的PR 疫情中,为我国根除PR 创造条件。
1 材料与方法
1.1 病毒、细胞和质粒PRV NY 株由郑州市猪重大疫病防控重点实验室分离、鉴定并保存;猪睾丸(ST)细胞购自中国兽药监察所;PRV 转移质粒p G由郑州市猪重大疫病防控重点实验室构建,其中(PstⅠ-PstⅠ)部分含有完整的绿色荧光蛋白基因(EGFP)表达盒,包括CMV、EGFP、MCS、SV40 Poly A 尾基因片段,大小约为1 700 bp;载体p MD 18-T 和p UC19购自宝生物工程(大连)有限公司。
1.2 主要试剂2×Taq Master Mix酶和DNA 凝胶回收试剂盒购自康为世纪生物技术有限公司;T4DNA 连接酶、限制性内切酶(Bam HⅠ、PstⅠ、HindⅢ)以及XfectTMTransfection Reagent购自宝生物工程(大连)有限公司;UNIQ-10 柱式病毒基因组抽提试剂盒、质粒小量提取试剂盒购自生工生物工程(上海)股份有限公司;OMEGA E.Z.N.A.TMEndo-Free Plasmid Mini KitⅠD6948-01B购自OMEGA 公司;DMEM 购自武汉博士德生物公司;2×DMEM、低熔点琼脂糖购自索莱宝科技有限公司。
1.3 转移质粒构建技术过程见图1。
1.4 引物设计与合成基于PRV 经典疫苗株Bartha-K61 株的基因缺失部位[13]和PRV TJ 株(KJ789182)US区基因序列,设计2 对特异性引物gEIL/F和gEIL/R、gEIR/F 和gEIR/R(表1),分别用于扩增左右同源臂。其中,左同源臂(L)包括部分gD 基因和部分gI基因;右同源臂(R)包括部分gE基因、完整的Us9基因和部分Us2基因。针对左右同源臂,设计了1对特异性引物gEIQS/F和gEIQS/R,用于gE、gI基因缺失部分的PCR 鉴定。此外,设计1对特异引物gB/F和gB/R,扩增gB全基因,用于鉴定重组病毒r PRV NY-gE-/gI-。引物均由生工生物工程(上海)股份有限公司合成。
1.5 左右同源臂的扩增与鉴定参照UNIQ-10柱式病毒基因组抽提试剂盒说明书,提取PRV NY 株基因组。利用2对特异性引物gEIL/F和gEIL/R、gEIR/F和gEIR/R,PCR 扩增左右同源臂。回收的PCR 产物,分别与p MD18-T 载体连接,得到重组质粒p MD-g E/gIL 和p MD-g E/gIR,转化入感受态细胞DH5α。经鉴定为阳性的重组质粒,送生工生物工程(上海)股份有限公司进行序列测定。通过NCBI Blast比对,以确定左右同源臂序列的正确性。
1.6 转移质粒pUC-gE/gILR 和p UC-gE/gILRE 的构建用限制性内切酶Q.Cut Hind Ⅲ和Q.Cut PstⅠ,分别对重组质粒p MD-g E/gIL 和p UC19载体进行酶切。用T4DNA 连接酶,将回收纯化的gE/gIL与线性化的p UC19进行连接,构建重组质粒p UC-g E/gIL,转化入感受态细胞DH5α,并进行酶切鉴定。用Q.Cut Bam HⅠ和Q.Cut PstⅠ分别对重组质粒p MD-gE/gIR 和p UC-g E/gIL 进行酶切,回收纯化,用同样的方法构建重组质粒p UCgE/gILR,转化入感受态细胞DH5α。用Hind Ⅲ、PstⅠ对转移质粒p UC-gE/gILR 进行酶切鉴定后,保存于-20℃备用。

表1 扩增同源臂所需引物
用Q.Cut PstⅠ对pG 质粒进行酶切,回收纯化的EGFP基因表达盒与同样处理的转移质粒p UCg E/gILR 进行连接,构建重组质粒p UC-gE/gILRE。用PstⅠ对重组转移质粒p UC-gE/gILRE 进行酶切鉴定,并保存在-20℃条件下以供后续实验使用。
1.7 rPRV NY-gE-/gI--EGFP+双基因缺失株的构建
1.7.1 转染 当细胞融合程度达到70%左右时,参照宝生物工程(大连)有限公司的XfectTMTransfection Reagent试剂说明书进行PRV NY 株基因组与转移质粒p UC-gE/gILRE 的共转染。约4 h后,移除含有转染试剂的培养液,换为2 m L 新鲜的含有10%胎牛血清的DMEM 培养液,放置培养箱继续培养。约48 h 后可冻融收取病毒液,3 000 r/min离心5 min后取上清至-80℃保存。
1.7.2 重组病毒的空斑纯化与鉴定 用无血清和无双抗的DMEM 细胞培养液对上述病毒液进行10倍系列稀释,将10-2~10-7稀释度的病毒液分别接种到ST 细胞90%融合的6孔细胞培养板中,然后用低熔点营养琼脂铺板进行重组病毒的蚀斑筛选,挑选在荧光显微镜蓝光下观察到的最大绿色荧光蚀斑。将收取的病毒液反复冻融3次后,重复上述操作,直至在蓝光条件下观察到的所有病毒蚀斑均可呈现绿色荧光。将得到的病毒命名为r PRV NYgE-/gI--EGFP+。利用特异性引物gEIQS/F 和g EIQS/R,对重组病毒r PRV NY-gE-/gI--EGFP+进行PCR 鉴定。PCR 反应参数:95℃5 min;94℃30 s,53℃30 s,72℃3 min,共进行35个循环;72℃终延伸10 min,4℃10 min。
1.8 rPRV NY-gE-/gI-双基因缺失株的构建按照1.7.1的方法进行r PRV NY-gE-/gI--EGFP+株基因组与转移质粒p UC-g E/gILR 的共转染。约48 h 后可冻融收取病毒液,3 000 r/min离心5 min后取上清至-80℃保存。按照1.7.2的方法进行重组病毒的空斑纯化。将得到的病毒命名为r PRV NY-gE-/gI-。同1.7.2,对重组病毒rPRV NYgE-/gI-进行缺失部分的PCR 扩增并测序,以验证r PRV NY-gE-/gI-中gE、gI双基因的缺失。再用gB全基因特异引物gB/F 和gB/R,对r PRV NYg E-/gI-进行gB基因的扩增。
1.9 重组病毒r PRV NY-gE-/gI-的生长特性将以10μL(106.0TCID50/0.1 m L)剂量的重组病毒r PRV NY-gE-/gI-、弱毒疫苗Bartha-K61 株以及亲本株PRV NY,分别接种于单层ST 细胞的24孔板中。接种病毒4 h后,每隔2 h收取细胞及其上清培养液。于-80℃条件下反复冻融3 次后,进行TCID50值的测定,根据结果绘制出病毒的一步生长曲线,并进行分析。
2 结果
2.1 左右同源臂的扩增与测序结果利用2对特异性引物gEIL/F 和g EIL/R、g EIR/F 和gEIR/R,分别进行PCR 扩增。PCR 产物经电泳后显示,分别扩增出了1 500,750 bp大小的片段(图2),与预期结果相吻合。测序结果显示,左、右同源臂片段大小分别为1 364,760 bp。经NCBI Blast比对,结果显示左、右同源臂与PRV TJ株相应基因区段的同源性均高达99.9%,表明成功获得同源臂序列。

图2 同源臂的扩增 M.DL2000 DNA Marker;1.左同源臂(g EL);2.右同源臂(g ER);3.阴性对照
2.2 转移质粒的酶切鉴定转移质粒p UC-gE/gILR 经HindⅢ、PstⅠ进行酶切,电泳结果出现2条带:一条带为左同源臂,大小约为1 500 bp;另一条带为p UC-19载体与右同源臂,大小约为3 500 bp(图3),与预期结果相符。
转移质粒p UC-gE/gILRE经PstⅠ进行酶切,电泳结果出现2条带:一条带为插入的含EGFP 基因的(PstⅠ-PstⅠ)酶切片段,大小约为1 700 bp;另一条带,包括左、右同源臂和p UC-19载体,大小约为5 200 bp(图3),与预期片段大小相符。
2.3 r PRV NY-gE-/gI--EGFP+的拯救及PCR 鉴定结果PRV NY 株基因组与转移质粒p UC-gE/gILRE进行共转染ST 细胞48 h后,收获转染液,经10倍系列稀释进行蚀斑筛选。每次纯化时,挑取最大稀释倍数孔中的最大绿色荧光蚀斑(图4)。随着纯化次数的增加,绿色荧光蚀斑逐渐增大,直至在蓝光下所有蚀斑均可呈现绿色荧光。经5轮蚀斑纯化,所有病毒蚀斑均可呈现绿色荧光,表明重组病毒已纯化完全。

图3 转移质粒的酶切鉴定图 M.DL5000 DNA Marker;1.p UC-g E/gILR 的HindⅢ/PstⅠ酶;2.p UC-g E/gILRE的PstⅠ酶切
提取r PRV NY-gE-/gI--EGFP+基因组,利用特异性引物g EIQS/F 和gEIQS/R,对左右同源臂间的缺失部分进行PCR 鉴定。结果显示,扩增出了1条约1 800 bp大小的片段(包括160 bp和EGFP基因表达盒),与预期结果相符(图5);表明成功拯救获得可表达绿色荧光蛋白的双基因缺失株rPRV NY-gE-/gI--EGFP+。
2.4 rPRV NY-gE-/gI-的拯救及PCR 鉴定结果
r PRV NY-gE-/gI--EGFP+基 因 组 与 转 移 质 粒p UC-g E/gILR 进行共转染ST 细胞48 h后,收获病毒液,经10倍系列稀释进行蚀斑纯化重组病毒。每次挑选最大稀释倍数孔中不呈现绿色荧光的蚀斑(图6),随着纯化次数的增加不呈现绿色荧光的蚀斑数量逐渐增加,直至第5次纯化时,所有蚀斑均不呈现绿色荧光,即成功拯救并纯化获得了去除EGFP基因的双基因缺失株rPRV NY-g E-/gI-。

图4 不同光照下观察到的蚀斑照片 A.常光下rPRV NY-gE-/gI--EGFP+形成的蚀斑;B.蓝光下rPRV NY-gE-/gI--EGFP+形成的蚀斑

图5 gE/gI缺失部分PCR 鉴定结果 M.DL5000 DNA Marker;1.r PRV NY-gE-/gI- 的 缺 失 鉴 定 结 果;2.rPRV NY-gE-/gI--EGFP+的缺失鉴定结果;3.阴性对照
提取rPRV NY-gE-/gI-基因组,利用特异性引物gEIQS/F 和gEIQS/R,对其左右同源臂间的缺失部分进行鉴定。结果显示,扩增出了1 条约160 bp大小的片段(图5)。测序结果显示,所测序列长度为160 bp,与预期结果相符。与PRV NY 株gE、gI基因序列进行比较,结果显示r PRV NYg E-/gI-双基因缺失株缺失区域长2 586 bp片段(包括gI基因3端913 bp、gE基因5端1 570 bp及gI与g E之间的序列103 bp),并在缺失部位留下了1个PstⅠ位点,与预期结果符合。
利用gB 全基因引物,以重组病毒rPRV NYgE-/gI-DNA 为模板,PCR 扩增gB基因。结果显示,扩增出了1条约3 000 bp大小的片段(图7),与预期结果相符。
2.5 重组病毒rPRV NY-gE-/gI-的生长特性结果
根据各个时间点的TCID50测定结果绘制一步生长曲线,如图8所示。双基因缺失毒株r PRV NYgE-/gI-和弱毒疫苗Bartha-K61 株,与亲本病毒PRV NY 株具有非常相似的生长曲线,但体外生长动力学比亲本毒株弱:在感染细胞后10~14 h,亲本株PRV NY 株的增殖速度极快,而双基因缺失毒株r PRV NY-gE-/gI-和弱毒疫苗Bartha-K61株的增值速度和病毒滴度均低于亲本毒株。

图6 不同光照下观察到的蚀斑照片 A.常光下rPRV NY-g E-/gI-形成的蚀斑;B.蓝光下rPRV NY-g E-/gI-形成的蚀斑
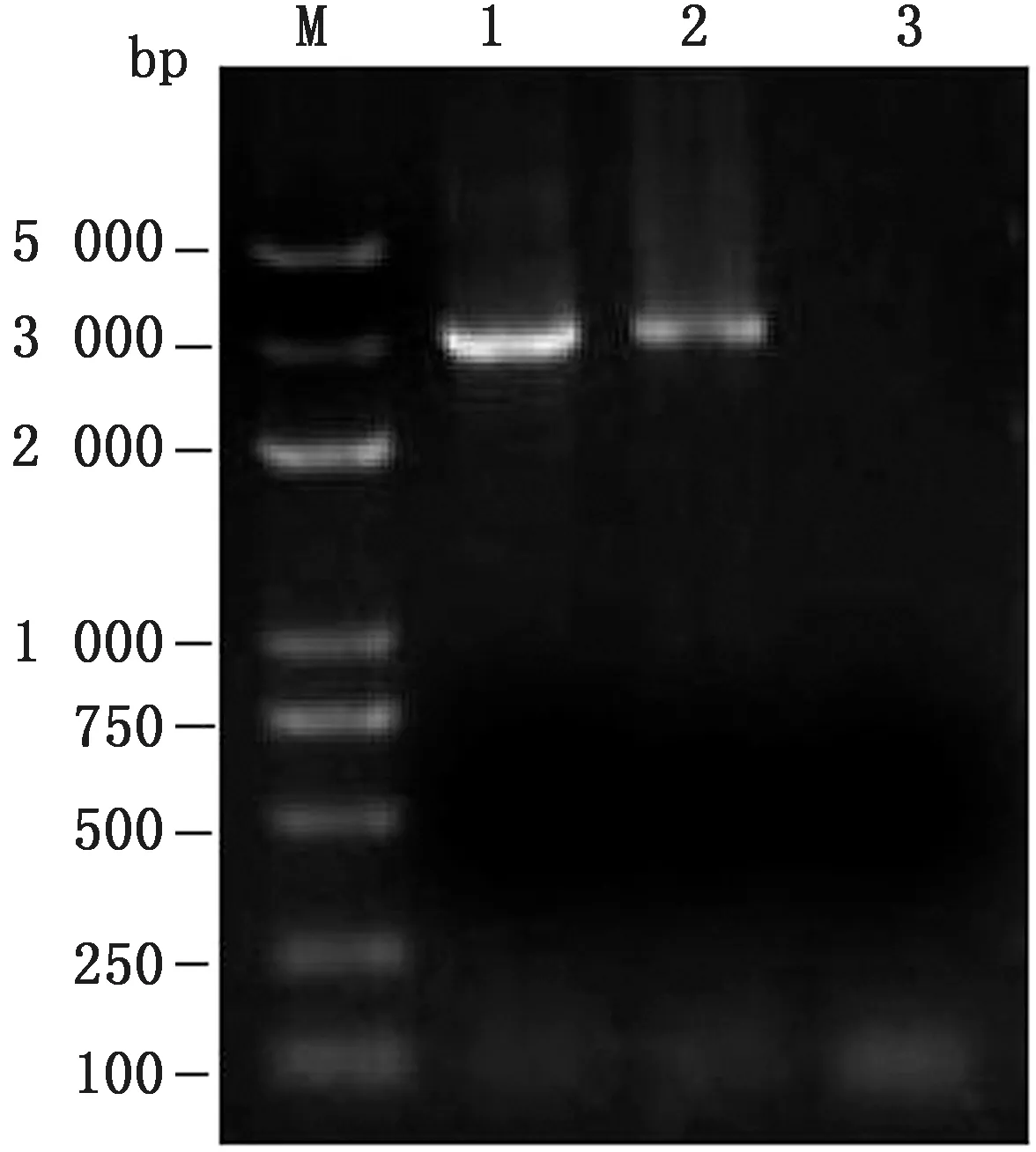
图7 重组病毒rPRV NY-gE-/gI-gB 基因的鉴定 M.DL5000 DNA Marker;1,2.rPRV NY-gE-/gI-的gB基因鉴定结果;3.阴性对照

图8 缺失病毒和亲本毒株的一步生长曲线
3 讨论
2011年,本实验室从河南省南阳市某猪场免疫过PRV Bartha-K61疫苗的病死猪体内分离到1株PRV 野毒株,命名为PRV NY 株。PRV NY 株对小鼠具有极强的致死性,与PRV Bartha-K61 株具有较大的抗原差异性;此外,基于g E、gB、gC基因的同源性和遗传进化分析结果显示,该毒株与国内近年分离的PRV 流行株同源性最高,且在进化树中与国内近年来分离的PRV 流行株位于同一个分支,因此我们以PRV NY 株为亲本株,进行PRV 基因缺失弱毒株的拯救。
gE和gI基因是PRV 的2个主要毒力基因,且均为PRV 复制的非必需基因,两者以共价键形式结合成复合体gE/gI从而参与PRV 在神经系统的侵袭和扩散[14-15]。PRV 的毒力受多种基因的共同控制,研究也证实若仅缺失gE 基因,则疫苗株仍会保留一定程度的毒力[16]。而将g E 和gI基因共同缺失会使PRV 的毒力在很大程度上得到减弱,同时可以对PRV 的神经嗜性造成影响[17-18]。现在临床上应用的PRV 基因缺失疫苗普遍都缺失了gE、gI基因,因此临床检测也普遍结合使用gE-ELISA 诊断方法来实现PRV 疫苗毒和野毒的高效鉴别。
同源臂的适当长度是保证同源重组顺利进行的前提。如果同源序列太短,其特异性就相对较低,则极有可能导致同源重组的结果混乱,而同源臂太长也会降低重组效率[19]。研究表明同源重组技术应用于构建重组病毒时,常用的左右同源臂片段大小约为1 kb[20]。王春花[21]构建PRV 基因缺失病毒株时,所采用的左右同源臂长度分别为1 313,1 055 bp。本研究构建PRV 基因缺失毒株时所采用的左右同源臂长度分别为1 364,760 bp,完全可以满足需要。通过转移质粒p UC-gE/gILRE和p UC-gE/gILR 分别与PRV 基因组的2次同源重组,结合反向筛选和蚀斑纯化,我们成功获得了不表达绿色荧光蛋白的rPRV-gE-/gI-双基因缺失株,其缺失片段为2 586 bp大小(含gE、gI基因),并在缺失部位留下了1个PstⅠ位点。由于本研究通过反向筛选去除了外源的EGFP基因,因此接种动物后不会诱导机体产生针对外源蛋白的抗体,大大提高了本缺失病毒的稳定性和生物安全性[18]。此外,在提取DNA 的过程中所有操作要求尽量轻柔,避免DNA 分子出现机械性破碎,影响共转染的重组效率。因此,我们采用柱式病毒基因组抽提试剂盒对PRV NY 株的基因组进行提取,该试剂盒操作过程比酚氯仿抽提法简便,且能获得纯度较高的病毒基因组,可获得尽可能多的完整的DNA 分子,为后续基因缺失病毒的拯救打下坚实的基础。
目前将携带外源基因的转移质粒导入真核细胞的技术方法有很多,例如最常用的脂质体转染法、电转化方法等,但大多转染试剂对细胞毒性强、损伤大,会造成细胞的大量死亡,而良好的细胞状态直接影响到病毒重组效率的高低。本研究使用了一种新型的可生物降解的纳米颗粒转染试剂XfectTMTransfection Reagent进行转染操作,该试剂是基于可降解的纳米颗粒制备的,对细胞毒性非常低,转染效率高,且在转染过程中兼容血清,无需使用OPTIMEM 培养液,操作简便。在共转染试验过程中,我们观察到细胞的生长始终处于较为良好的状态,为重组病毒的成功拯救提供了良好的环境。
该研究下一步还将对该疫苗候选株的遗传稳定性、对靶动物和非靶动物的安全性、在妊娠母猪和新生仔猪上的免疫效力以及免疫持续期等进行系统评价,以期望将其用于当前PR 新疫情的防控工作,为我国根除PR 创造条件。
