超高效液相色谱-串联质谱法测定茶叶中草甘膦和草铵膦的残留量
2019-06-11刘文锋王安波潘承丹
杨 梅,孙 思,刘文锋,王安波,潘承丹,汪 俭*
(黔东南州农产品质量安全检测中心,贵州 凯里 556000)
草甘膦是世界上应用最广、生产量最大的除草剂,其作为无选择类除草剂,以其高效、低毒、廉价等特性被广泛运用于全球各个农业和非农业领域[1]。草铵膦可以有效去除绝大部分耐草甘膦杂草,且具有环保易降解等特点而受到各界青睐。近年来由草甘膦引起的水体污染、植被破坏、牲畜中毒等案件持续增加[2-3]。随着其在茶叶上的运用日益加剧,其残留问题也越来越受关注,为此,世界各国均对茶叶中草甘膦和草铵膦的最大残留限量制定了严格的规定。欧盟规定茶叶中草甘膦[4]和草铵膦的最大残留限量分别为2.0 mg/kg和0.1 mg/kg,日本肯定列表制度对茶叶中草甘膦和草铵膦的最大残留限量为1.0 mg/kg和0.3 mg/kg,我国GB 2763—2016《食品中农药最大残留限量》规定茶叶中草甘膦和草铵膦的最大残留限量分别为1.0 mg/kg和0.5 mg/kg。
草甘膦和草铵膦的结构类似,具有强极性、难溶于各种有机溶剂的特点,这也就决定了对其残留量进行准确定量分析难度较大,目前主要的检测方法有免疫技术法[5-6]、气相色谱法[7-8]、高效液相色谱法[9]、离子色谱法[10-13]、毛细管电泳法[14]、气相色谱-质谱联用法[15-16]、高效液相色谱-质谱联用法等[17-22],但大部分方法存在灵敏度低、操作程序繁琐、有机试剂污染严重等缺点,也难以直接运用于茶叶农残检测领域。目前国内外利用超高效液相色谱-串联质谱法同时检测茶叶中草甘膦和草铵膦残留的方法报道较少,我国目前尚无茶叶中检测草铵膦的国家标准,而GB 2763—2016中所推荐的茶叶中草甘膦的检测标准SN/T 1923—2007《进出口食品中草甘膦残留量的检测方法 液相色谱-质谱/质谱法》实际运用时步骤繁琐、耗时长,回收率不稳定,定量限偏高。因而亟待建立一种实用且快速检测草甘膦和草铵膦的分析方法。考虑到超高效液相色谱法-串联质谱法的高灵敏度、高效率、精确性[23-24],本研究采用NaOH溶液提取样品中的草甘膦和草铵膦残留,提取液经氯甲酸(9-芴甲基)酯(9-fluorenylmethyl chlorformate,FMOC-Cl)衍生反应后,用超高效液相色谱-三重四极杆质谱联用仪测定,采用外标法定量,建立茶叶中草甘膦和草铵膦药物残留的快速检测方法。
1 材料与方法
1.1 材料与试剂
茶叶 市购;草甘膦(C5H18N3O4P,CAS号:77182-82-2,纯度≥99.3%)、草铵膦(C5H18N3O4P,CAS号:1071-83-6,纯度≥99.3%) 北京坛墨质检科技有限公司;FMOC-Cl(纯度≥99%) 北京百灵威科技有限公司;甲醇、乙腈(均为色谱纯) 美国Tedia公司;甲酸铵(质谱纯) 美国Fisher Scientific公司;N-丙基乙二胺(N-(n-propyl) ethyenediamine,PSA,粒度40 μm) 美国Supelco Analytical公司;氢氧化钠(分析纯) 成都金山化学试剂有限公司;盐酸(36%~38%,优级纯) 西陇科学股份有限公司;硼酸钠(Na2B4O7•10H2O,纯度≥99.5%) 天津市永大化学试剂有限公司;超纯水。
1.2 仪器与设备
UPLC H-Class/TQ-S Micro超高效液相色谱-三重四极杆质谱联用仪 美国Waters公司;氮气发生器 英国Peak公司;H2050R高速冷冻离心机 湖南湘仪实验室仪器开发有限公司;SCQ-151104E超声波清洗器 上海声彦超声波仪器有限公司;SHA-C水浴恒温振荡箱江苏科析仪器有限公司;DH500B II电热恒温培养箱天津市泰斯特仪器有限公司;VORTEX2涡旋振荡仪德国IKA公司。
1.3 方法
1.3.1 标准溶液配制
草铵膦、草甘膦标准储备溶液(1.0 mg/mL):分别精确称取草铵膦、草甘膦标准品各1 0.0 mg于10 mL容量瓶中,用水溶解后定容10 mL。放置于0~4 ℃冰箱,有效期6 个月。
草铵膦、草甘膦标准中间工作溶液(10.0 μg/mL):分别吸取0.1 mL草铵膦、草甘膦标准储备溶液于10 mL容量瓶中,用水定容至10 mL。放置于0~4 ℃冰箱,有效期1 个月。
草铵膦、草甘膦混合标准工作溶液(1.0 μg/mL):分别吸取1.0 mL草铵膦、草甘膦标准中间工作溶液于10 mL容量瓶中,用水定容10 mL,现用现配。
1.3.2 样品处理
1.3.2.1 样品制备
将茶叶样品放入粉碎机中粉碎,样品全部过425 μm的标准网筛,装入洁净的盛样容器内,密封并标明标记,于-18 ℃保存。
1.3.2.2 样品提取
精密称取(1±0.01)g试样,置于50 mL离心管中,加入10 mL 0.05 mol/L NaOH溶液,涡旋混匀1 min;室温水浴振荡提取20 min,经10 000 r/min离心5 min,上清液用滤纸过滤至10 mL离心管中得提取液I;取2 mL提取液I加入1 mol/L HCl溶液20 μL,涡旋混匀后10 000 r/min离心5 min得提取液II,将提取液II倒入预先称好0.2 g PSA的10 mL离心管中,并立即涡旋混匀,10 000 r/min离心5 min得提取液III。取1 mL的提取液III置于10 mL离心管中,加入0.2 mL的硼酸钠缓冲溶液(50 g/L),摇匀后再加入0.2 mL FMOC-Cl(20 g/L)衍生试剂,涡旋混匀振摇后置37 ℃静置过夜,衍生后的样品经10 000 r/min离心5 min后,过水系滤膜,供超高效液相色谱-串联质谱仪分析。
1.3.3 仪器条件
1.3.3.1 色谱条件
采用ACQUITY UPLC HSS T3色谱柱(2.1 mm×100 mm,1.8 μm);柱温40 ℃;样品温度20 ℃;流动相:A为5 mmol/L甲酸铵-甲醇溶液,B为5 mmo/L甲酸铵溶液;梯度洗脱模式;流速0.4 mL/min;进样量1 μL。洗脱程序参见表1。

表1 流动相梯度洗脱程序Table 1 Gradient elution program
1.3.3.2 质谱条件
采用电喷雾离子源;正离子扫描,多反应监测(multiple reaction monitoring,MRM),脱溶剂温度500 ℃,脱溶剂气氮气流速1 000 L/h,离子源温度150 ℃,锥孔气流量50 L/h,毛细管电压3.0 kV,碰撞气为氩气。 其他质谱参数见表2。
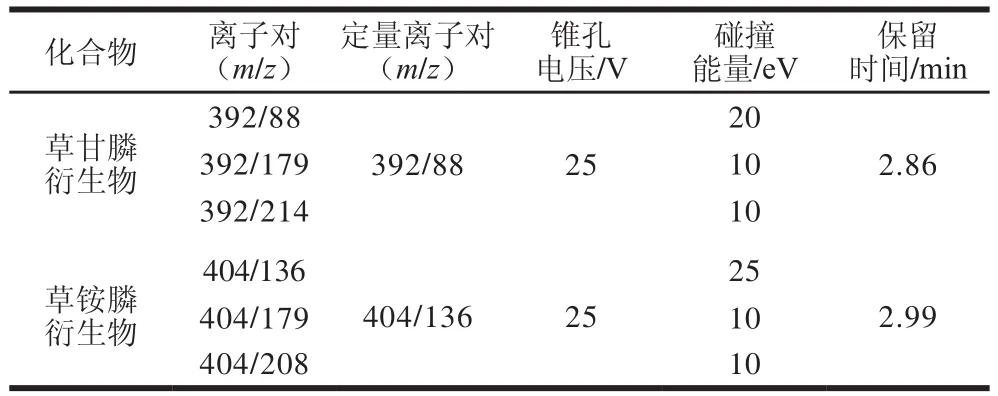
表2 质谱参数Table 2 Mass spectrometric parameters
1.4 数据分析
所有测试重复平行实验6 次,数据在Waters公司的MassLynx V4.1软件下采集,采用MassLynx V4.1SCN945处理方法对检测结果进行定性定量处理分析和并采用Origin 9.0进行图像处理。
2 结果与分析
2.1 前处理条件优化
2.1.1 提取溶剂和提取方式的选择
草甘膦和草铵膦极性较大且易溶于水,难溶于大多数有机溶剂,因此常采用强极性溶剂进行提取,如水[25]、水-二氯甲烷[19]、0.2%甲酸-水(0.2∶99.8,V/V)[26]和氢氧化钾[4]等,本研究比较水、水-二氯甲烷、0.2%甲酸-水、0.05 mol/L氢氧化钾、0.05 mol/L氢氧化钠5 种溶剂提取,每种提取溶剂进行6 个平行实验,以平均值作为结果进行判断;结果表明氢氧化钠提取效果最好,可能是在碱性环境下,草甘膦等会反应生成效应的盐,可提高其溶解率,而且茶多酚更易氧化生成沉淀,已达到净化的目的[4]。因此提取试剂选择氢氧化钠溶液提取,见图1。
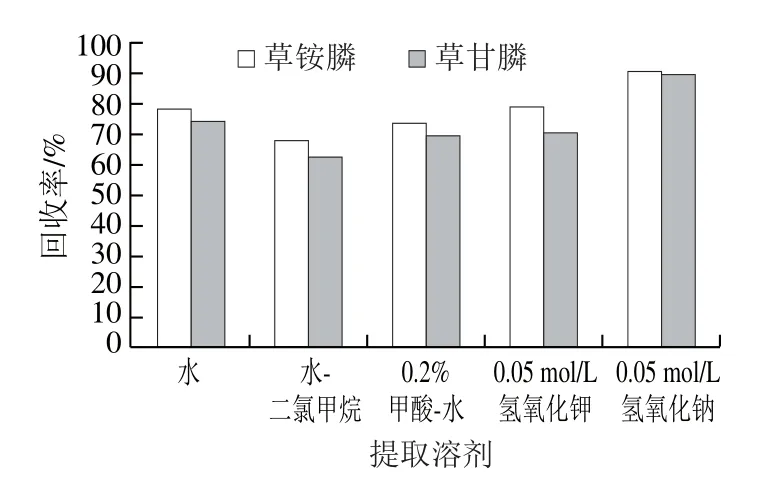
图1 提取溶剂的选择Fig. 1 Selection of extraction solvent
提取效率方面,采用空白样品加标(1 mg/kg)的方法比较超声提取、静置提取和振荡提取3 种提草甘膦和草铵膦取方式的提取效果,每种提取方法进行6 个平行实验,取平均值作为结果判断,结果表明提取效率振荡提取>超声提取>静置提取;此外在振荡提取的前提下,采用不同浓度的氢氧化钠溶液(0.01~0.1 mol/L)提取样品,0.05 mol/L氢氧化钠提取效果最佳,见图2。
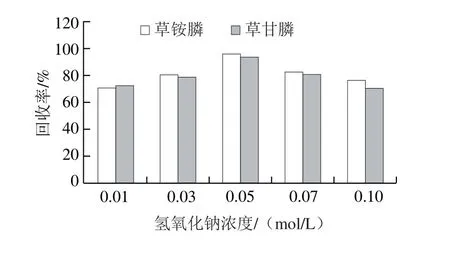
图2 提取溶剂氢氧化钠溶液浓度优化Fig. 2 Optimization of NaOH concentration
2.1.2 HCl浓度的优化
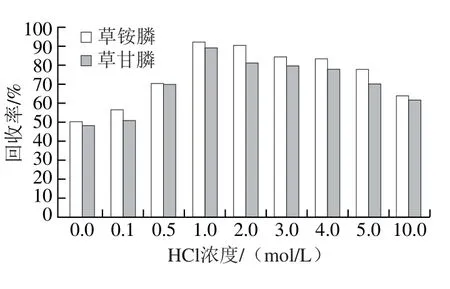
图3 HCl溶液浓度的优化Fig. 3 Optimization of HCl concentration
采用空白样品加标(1 mg/kg)的方式,样品采用0.05 mol/L氢氧化钠溶液提取,取提取液I 2 mL于10 mL离心管中,加入20 μL 0~10 mol/L不同浓度HCl溶液调节酸度,按照1.3.2节提取,每个浓度重复6 次,取平均值作为结果进行判断。如图3所示,提取液I未加HCl溶液时,回收率在50%左右,同时杂峰相对较多,而当HCl溶液添加浓度为1 mol/L时,回收率最高,在92%左右。原因可能是HCl打破了原有茶汤中的电解质平衡,在离子效应、电解质作用和共沉效应等共同作用下,茶多酚、氨基酸、糖类等聚合成大分子物质形成络合物而沉淀净化效果提高[27]。因此选择1 mol/L HCl溶液。
2.1.3 净化条件优化
样品0.05 mol/L氢氧化钠提取,经1 mol/L HCl溶液处理后,取2 mL提取液II空白加标(0.1 μg/mL),经不净化、0.2 g PSA、0.2 g C18净化粉、QuEChERS法、HLB固相萃取柱、C18固相萃取柱净化,按照1.3.2节方法衍生,每种净化条件进行6 个平行实验,取平均值作为结果判断,结果如图4所示。样品经0.2 g PSA净化后,有效去除样品中的色素以及杂质,其回收率较其余几种净化效果好,回收率达95%左右,净化效果最好且操作简单、节省时间,适合大批量样品检测。因此采取0.2 g PSA对样品进行净化。
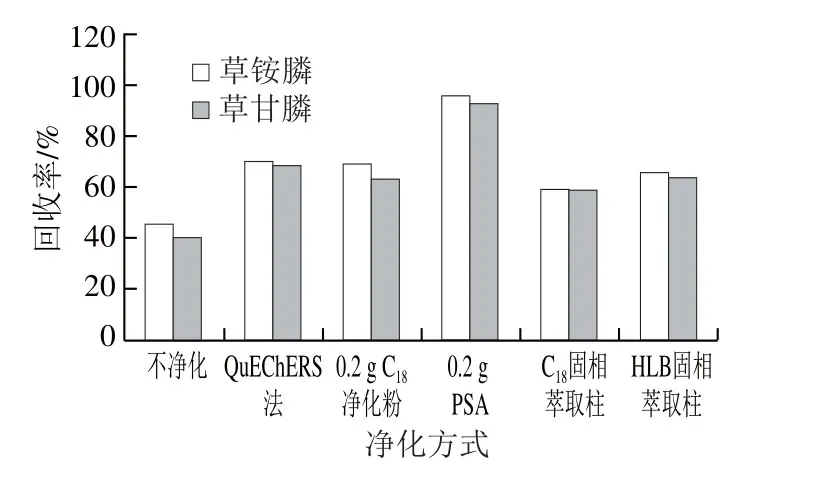
图4 净化条件选择Fig. 4 Selection of clean-up sorbent
空白样品经1.3.2节方法提取后,得提取液II,采用空白提取液加标(0.1 μg/mL)的方式进行,添加0~0.5 g PSA,净化衍生后上机检测。比较不同用量PSA对净化效果的影响,结果如图5所示。当用量为0.2 g时,回收率最高,94%左右,继续添加用量则无明显变化,因此PSA用量选择0.2 g。
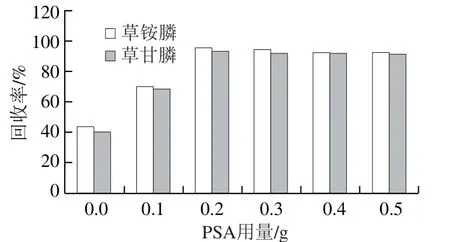
图5 PSA用量优化Fig. 5 Optimization of PSA dosage
2.1.4 衍生条件优化
空白样品经1.3.2节发法提取后,得提取液III,采用空白提取液加标(0.1 μg/mL)的方式进行,添加0.2 mL 20~100 g/L硼酸钠缓冲溶液,加入0.2 mL FMOC-Cl(20 g/L)衍生后上机检测,比较不同浓度硼酸钠缓冲溶液对衍生效果的影响,每种衍生条件进行6 个平行实验,取平均值作为结果判断,结果如图6所示。当添加质量浓度为50 g/L时,回收率最高,96%左右。同样采用空白提取液加标(0.1 μg/mL)的方式,加入0.2 mL 50 g/L硼酸钠缓冲溶液,再加入质量浓度5~50 g/L FMOC-Cl衍生试剂,比较不同浓度FMOC-Cl对衍生效果的影响,结果如图7所示,衍生试剂质量浓度为20 g/L时,衍生效果最好,回收率达96%左右。
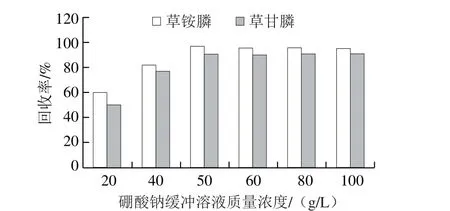
图6 硼酸钠缓冲溶液质量浓度优化Fig. 6 Optimization of concentration of Na2B4O7·10H2O
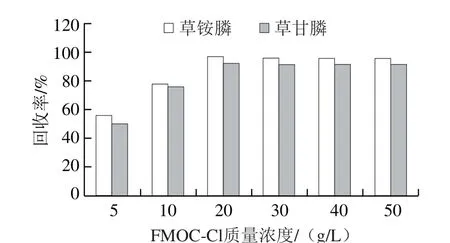
图7 FMOC-Cl质量浓度优化Fig. 7 Optimization of FMOC-Cl concentration
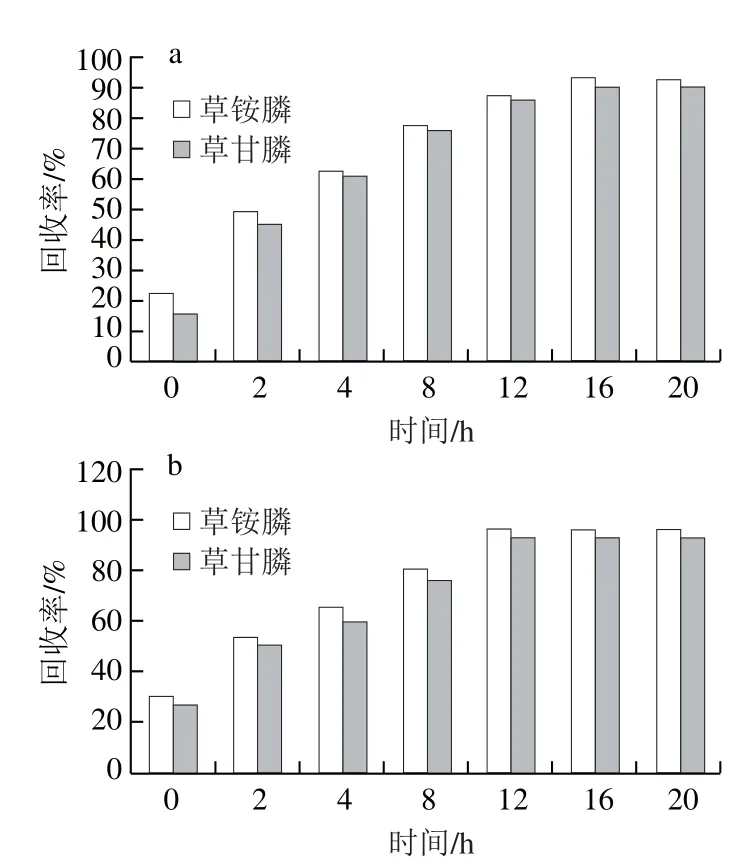
图8 25 ℃(a)和37 ℃(b)条件下衍生时间的优化Fig. 8 Optimization of derivatization time at 25 (a) and 37 ℃(b)
提取液加标(0.1 μg/mL)的方式进行,比较不同衍生时间对衍生效果的影响。分别比较37 ℃衍生2、4、8、12、16、20 h和25 ℃衍生2、4、8、12、16、20 h,每个衍生时间进行6 个平行实验,取平均值作为结果判断,结果如图8所示,结果表明37 ℃衍生过夜(12 h以上)效果最好,回收率达95%以上,之后时间增长也无明显增加,因此衍生时间为37 ℃衍生过夜(12 h以上)。
2.2 色谱条件优化
2.2.1 色谱柱的选择
分别采用Waters ACQUITY UPLC BEH C8、Waters ACQUITY UPLC BEH C18、Waters ACQUITY UPLC HSS T3进行分析,发现Waters ACQUITY UPLC BEH C8不适合草甘膦和草铵膦的分析;采用Waters ACQUITY UPLC BEH C18分析时,目标峰与杂峰出现部分重叠,分离差,且存在拖尾比较严重,因此也不适用于草甘膦和草铵膦的分析;而采用Waters ACQUITY UPLC HSS T3分析时,目标峰与杂峰完全分离,峰形较好,无拖尾现象,因而选择Waters ACQUITY UPLC HSS T3进行分析。
2.2.2 流动相的优化
此外,还比较甲醇-水、甲醇-5mmo/L甲酸铵溶液、乙腈-水、乙腈-5 mmo/L甲酸铵溶液、甲醇-5mmo/L甲酸铵溶液、乙腈-5mmo/L甲酸铵溶液、5 mmol/L甲酸铵/甲醇-5 mmo/L甲酸铵溶液、5 mmol/L甲酸铵/乙腈-5 mmo/L甲酸铵溶液对目标物的色谱峰及离子化的影响,结果发现5 mmol/L甲酸铵/甲醇-5 mmo/L甲酸铵溶液作为流动相对2种化合物的分离度较好,峰形较好,且目标化合物的响应较高,于溶剂中加甲酸铵有利于目标化合物离子化,从而提高灵敏度,所以选择5 mmol/L甲酸铵/甲醇-5 mmo/L甲酸铵溶液作为流动相。
2.3 质谱条件优化
将2 种药物用超纯水分别配成300 μg/L的标准溶液1.0 mL,依次加入0.2 mL硼酸钠缓冲溶液(50 g/L)和0.2 mL FMOC-Cl(20 g/L)进行衍生,衍生后经高速冷冻离心机离心后过0.22 μm滤膜,不经色谱柱,采用电喷雾离子源的正离子模式扫描,MRM模式,直接质谱进样,进行母离子扫描和子离子扫描,通过自动和手动,调谐相结合,优化二级碎片离子信息,优化锥孔电压、碰撞能量,获得定性离子和定量离子最佳质谱条件,具体见表2。
2.4 基质效应和标准曲线绘制
基质效应(matrix effect,ME)指样品中的其他成分对待测物测定值的影响,即基质对分析方法准确测定分析物的能力的干扰。根据基质对检测信号响应值的不同影响,基质效应可分为基质增强效应和减弱效应。由于草甘膦和草铵膦的极性很大,实验采取碱溶液进行提取,提取过程中被提取出来的有机或无极分子(如无机盐、酚类、色素和脂类),还有前处理过程中引入的缓冲盐、衍生剂和溶解液等[19]。采用提取后添加法[28],通过比较以水为溶剂和以空白提取液为溶剂分别配制0.005、0.010、0.050、0.10、0.20、0.30、0.40、0.50 μg/mL的配制系列标准溶液,按照1.3.2节处理,以标准溶液质量浓度为横坐标,峰面积为纵坐标,绘制标准曲线。比较基质与空白溶剂标准曲线的斜率,从而评估二者的基质效应。由表3可知,2种药物质量浓度在0.005~0.50 μg/mL范围内时,各标准曲线相关系数(R2)均大于0.995,见表3。0.10 μg/mL的纯水标液和空白基质衍生后的色谱图见图9。

表3 2 种化合物的线性方程、相关系数、加标回收率、相对标准偏差=10)和基质效应Table 3 Linear equations, correlation coeffificients, spiked recoveries,relative standard deviations (RSDs) and matrix effect for analytes ( = 10)
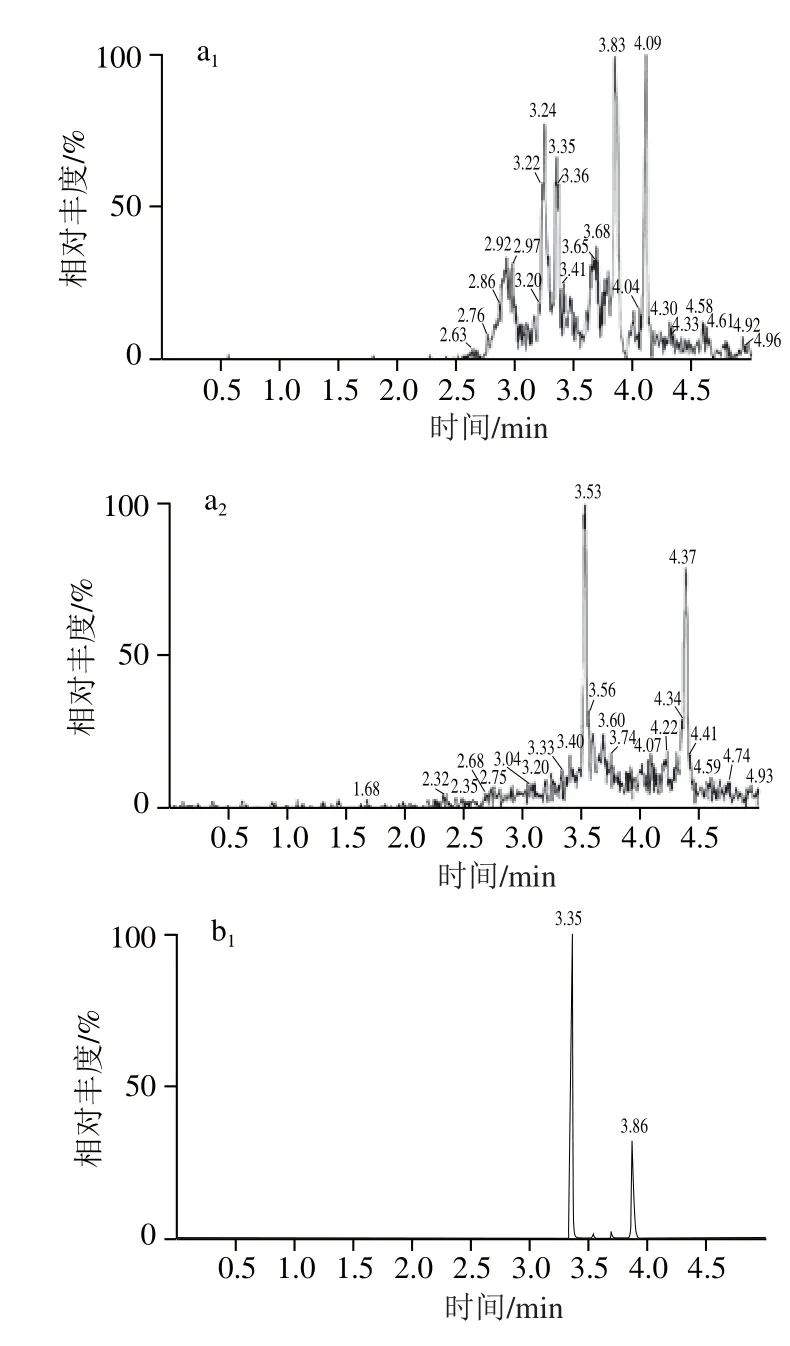
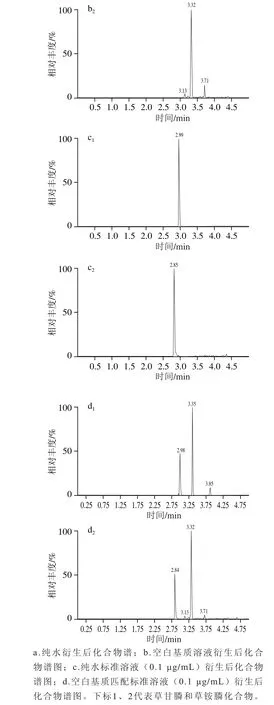
图9 化合物离子流色谱图Fig. 9 Total ion current chromatograms for GLY and GLUF from samples
基质效应/%=(空白基质标曲的斜率-溶剂标曲的斜率)/溶剂标曲的斜率×100[29-30]。│ME│<0,表明基质的存在抑制了目标化合物的响应值;ME=0,表明基质的存在对该目标化合物的响应无影响;ME>0,说明基质的存在增强了目标化合物的响应。基质效应按其绝值大小可分为3个等级[31-32]:│ME│>50%,为强基质效应;20%<│ME│≤50%,为中等强度基质效应;0%<│ME│≤20%,为弱基质效应。从基质标与溶剂标的响应相差很大,基质效应比较明显。从表3可以看出,草甘膦、草铵膦ME值<0,意味着基质的存在抑制了目标化合物的响应值;当添加量为1 mg/kg时,采用溶剂标曲计算该方法提取2 种化合物回收率在中为51.2%~70.7%,而基质匹配标曲计算该方法提取2 种化合物的回收率为88.2%~96.6%,可知草甘膦、草铵膦均为中等强度基质效应,因此采用基质匹配标准曲线进行定量测定。
2.5 准确度和精密度实验结果
采用基质匹配标准溶液-外标法定量,取茶叶空白样品基质(1±0.01)g,分别添加3 个水平的草甘膦和草铵膦化合物,加标量0.08、0.10、1.0、2.0 mg/kg和4.0 mg/kg,按照1.3.2节的方法进行操作,每个加标水平重复10 次,结果如表3所示。
从表3可以看出,绿茶空白基质样品在0.08、0.10、1.0、2.0 mg/kg和4.0 mg/kg 5 个添加量的草甘膦和草铵膦化合物,进行添加回收加标实验,草甘膦的回收率为75.6%~95.2%,相对标准偏差为3.24%~8.38%(n=10),草铵膦的回收率为76.0%~96.6%,相对标准偏差为3.05%~7.85%(n=10)。说明该方法对茶叶中草甘膦和草铵膦化合物药物残留的测定均有良好的准确度和精密度,符合残留分析的要求。
2.6 检出限和定量限测定结果
采用不含草甘膦和草铵膦的空白基质样品提取液逐级稀释草铵膦和草甘膦标准溶液,当定量离子对和定性离子对的信噪比为3时所对应的草铵膦和草甘膦的含量为0.05 mg/kg,当信噪比为10时,所对应的草铵膦和草甘膦含量为0.08 mg/kg,选择符合一定的准确度和精密度要求的最低浓度,采用加标回收进行验证。实验结果见表3,因此2 种药物的检出限为0.05 mg/kg;定量限为0.08 mg/kg。
2.7 样品测定结果
采用优化后的前处理方法和分析条件对所抽的200 批绿茶样品进行草甘膦和草铵膦药物残留检测,以保留时间、离子比率进行定性分析,以定量离子对进行定量分析。其中20 批样品有检出草甘膦含量依次为0.085~0.65 mg/kg,15 批样品检出草铵膦0.12~0.45 mg/kg,均未超过相关规定限量。
3 结 论
本研究是采用氢氧化钠溶液 提取样品中的草甘膦和草铵膦药物残留,HCl溶液调节pH值,经PSA净化,FMOC-Cl衍生化,正离子MRM测定,外标法定量。优化后的方法提取效率高、净化效果好,提高工作效率。方法能对茶叶中的草甘膦和草铵膦药物残留进行定性和定量分析,方法具有较高的回收率、良好的精密度、较高准确度,选择性好,操作快速、简便,满足实际工作需求。
