在线微固相萃取/高效液相色谱联用分析环境水样中磺胺类药物
2018-12-19周婵媛聂胜强
周婵媛,罗 军,王 壹,严 伟,聂胜强
(贵阳学院 化学与材料工程学院,贵州 贵阳 550005)
磺胺类药物(Sulfonamides,SAs)属于化学合成类抗生素,被广泛应用于防治人类、水产和畜禽养殖过程中由细菌感染引发的疾病。SAs的大量生产和广泛应用对环境造成了严重污染,尤其是水体的污染。而农作物吸收了被污染的水源并被人类食用后可能对人体健康形成危害。基于SAs污染造成的危害,亟需对污染水体进行处理,而现有的污水处理工艺仅针对废水中耗氧有机物和营养元素氮磷,对SAs污染物几乎无处理效果。因此,迫切需要建立有效、快捷、简便的样品前处理富集分析技术。目前SAs的检测方法主要有高效液相色谱法[1-3]、液相色谱-质谱联用法[4-5]、气相色谱-质谱联用法[6]、电化学检测法[7-8]等。然而,SAs具有强极性且在环境样品中含量较低,导致其分析检测难度增大。快速有效的样品前处理方法可对目标物进行有效的分离富集,是提高SAs检测灵敏度的重要途径。目前,国内外对SAs的前处理方法有液-液萃取法[9-10]、基质固相萃取法[11-12]和固相萃取法[13],但主要以离线方式进行,耗时长,操作复杂,且溶剂消耗量大。将样品前处理与分析仪器在线联用,实现前处理与分离分析的优化组合,可以提高分析的自动化程度,减少离线操作中的人为误差和样品损失,是当前复杂样品前处理分离分析的重要发展方向。
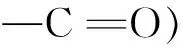
本文基于Sep的高比表面积、良好化学稳定性和热稳定性等优异性能,通过酸化法制备氧化海泡石(O-Sep)功能化材料,研究了O-Sep材料的结构和萃取性能,并建立了分析环境水样中磺胺对甲氧嘧啶(Sulfameter,SMD)、磺胺多辛(Sufladoxine,SDX)、磺胺苯吡唑(Sulfaphenazole,SPP)3种磺胺类药物的O-Sep在线微固相萃取/高效液相色谱(O-Sep/μ-SPE/HPLC)联用方法。
1 实验部分
1.1 仪器与试剂
SK2200H超声波清洗器(天津奥特赛恩斯仪器有限公司);101-1AB电热鼓风干燥箱(天津市泰斯仪器有限公司);ST16R离心机(美国Thermor公司);LC-10Avp 型高效液相色谱仪(日本 Shimadzu 公司),配备 CLASS-VP 工作站,SPD10AVP 紫外检测器,Diamonsil C18色谱柱(250mm×4.6mm i.d.,5μm,迪马公司);FA1604型电子天平(上海天平仪器厂);Quanta600电子扫描显微镜(SEM,荷兰 FEI 公司);NICOLET AVATAR330傅立叶红外光谱仪(美国热电公司);Orion868型酸度计(美国ThermoElectron公司)。
标准品:磺胺对甲氧嘧啶、磺胺多辛、磺胺苯吡唑(百灵威科技有限公司);海泡石原矿(湘潭九华碳素高科有限公司);丙酮、乙酸乙酯、正己烷(分析纯,天津大茂化学试剂厂);乙腈、甲醇(色谱纯,美国Tedia公司);实验用水为超纯水,由Millipore 纯化系统制备。其他试剂均为分析纯。
1.2 氧化海泡石的制备
以Sep原矿为原料,用酸化法制备O-Sep[22-24]:称取5.0g过200目筛的Sep粉末置于盛有50mL50%(体积分数)乙醇溶液的烧杯中,搅拌10min,减压抽滤,用水反复清洗。过滤后将收集的产物放入100mL4mol/L HCl溶液中,于80℃磁力搅拌8h。过滤,用超纯水反复洗涤,直至上清液接近中性,然后将样品分散在超纯水和乙醇的混合溶液(1∶1,体积比)中,超声(200W,24kHz)2h,使其充分分散,离心收集上清液,用超纯水洗涤5次,于105℃干燥,所得固体产物即为O-Sep。
1.3 微填充柱的制备
10mg O-Sep粉末填充于聚醚醚铜(PEEK)制成的2.0mm内径微柱中,柱两端用不锈钢筛板固定填料,然后将其装入相匹配的色谱保护柱的不锈钢套内。将填充柱连接于高压液相泵上,用乙腈(含1%甲酸)以0.05mL/min 冲洗48h,再用超纯水在0.1mL/min流速下冲洗4h,直至流出液接近中性。
1.4 固相萃取材料的表征
将O-Sep材料用导电胶固定,经喷金处理后,采用冷场扫描电子显微镜在100~10000的放大倍率下观察材料形貌与结构。采用X射线衍射仪研究O-Sep和Sep粉末的晶态结构。测定条件如下:电压40kV,电流30mA,扫描速度5°/min。将O-Sep材料与溴化钾混合均匀研磨,利用红外灯光照射干燥,在400~4000cm-1波数范围内测定红外吸收光谱。
1.5 萃取条件的研究
以pH3.0~7.0的磷酸缓冲溶液(20mmol/L)分别配制100μg/L的SAs混合标准溶液,用O-Sep/μ-SPE/HPLC在线联用装置,对萃取溶剂、萃取流速、解吸溶剂和解吸体积进行优化。
1.6 样品溶液的制备
将SMD、SDX和SPP先用乙腈配成1.0g/L的储备液,于4℃避光保存,再用pH6.0的磷酸缓冲溶液(20mmol/L)配成100μg/L的水溶液用于固相萃取条件的优化。以实验室自来水、鱼梁河水和贵阳市采集的4月份雨水为实际样品,以磷酸缓冲溶液(20mmol/L)调至pH6.0,经0.22μm滤膜过滤后存于4℃冰箱中备用。水样中加入对应体积的1.0mg/L 标准溶液,充分混匀,作为SMD、SDX和SPP的加标样品。后续处理步骤与不加标样品相同。
1.7 色谱条件
色谱柱:Dikma Diamonsil C18(250mm×4.6mm i.d.,5μm);流动相:有机相为乙腈,水相为0.1%(体积分数)甲酸水溶液。流速为1mL/min;梯度洗脱程序:0~20min,乙腈由10%线性升至40%,保持5min;紫外检测波长为270nm。
2 结果与讨论
2.1 固相萃取柱的结构与性能表征
通过扫描电子显微镜(SEM)研究了Sep和O-Sep的形貌(图1)。O-Sep通过酸化处理去除了表面的大部分杂质,呈现疏松多层结构,具有良好的通透性,且表面形貌发生了明显改变,与Sep相比表现出良好的分散性。O-Sep的比表面积增至169.38m2/g,为Sep的7.4倍。说明O-Sep良好的分散性提高了材料的比表面积,从而有利于增强萃取性能。

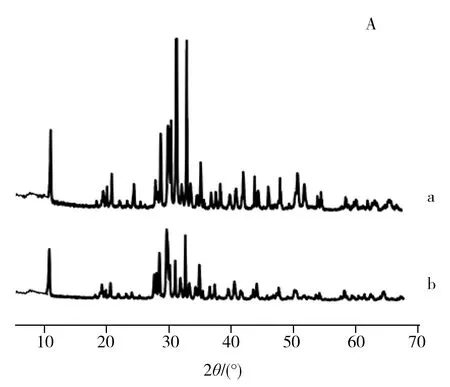
Sep和O-Sep的X射线衍射图谱如图2A所示,O-Sep在2θ=30.0°处对应于CaCO3的特征衍射峰强度与Sep相比明显降低,而在2θ=27.3°处的海泡石特征衍射峰更加明显,表明氧化处理去除了Sep中的CaCO3等杂质组分,提高了Sep的纯度。
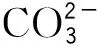
2.2 固相萃取条件的优化
2.2.1萃取溶剂采用 100 μg/L的SMD、SDX和SPP标准溶液考察了O-Sep固相萃取柱在不同pH值条件下的萃取效果。结果表明,当溶液pH值为6.0时,O-Sep固相萃取柱对SAs的吸附效率最好。随着pH值的增大,O-Sep固相萃取柱对SAs的吸附效率反而降低,这是由于溶液pH值影响了吸附剂的结构和磺胺类药物的形态,随着pH值的变化,SAs的存在形式不同,质子化程度不同。在pH 4.0~6.0条件下,目标物质子化呈电中性,O-Sep与目标物间的氢键作用促使O-Sep对SAs的萃取效果较好。当溶液pH值大于6.0时,SAs转变为阴离子形式,离子交换作用减弱,使得O-Sep对SAs的吸附量减少。综上,实验选择pH 6.0的磷酸缓冲液(20 mmol/L)为最优萃取溶剂。
2.2.2萃取流速保持过萃取柱的5.0 μg/L混合标准溶液体积为2.0 mL,考察了萃取流速(200、400、600、800 μL/min)对实验结果的影响。结果显示流速的改变对萃取结果影响不大,但流速为600 μL/min时萃取效果最佳,因此选择萃取流速为600 μL/min。
2.2.3解吸溶剂分别选择乙腈、0.1%氨水溶液-乙腈(5∶95) 、甲醇、0.1%氨水溶液-甲醇(5∶95)作为解吸溶剂进行优化,考察了解吸溶剂种类的影响。结果显示,在相同条件下,甲醇解吸体系对SAs的解吸效果明显高于乙腈解吸体系,且以0.1%氨水溶液-甲醇(5∶95)的效果最好,故选作解吸溶剂。
2.2.4解吸体积考察了不同解吸体积(200、300、400、500、600 μL)对目标物洗脱能力的影响。结果表明,随着解吸体积的增大,SMD、SDX和SPP的解吸量均逐渐增加。当解吸体积为500 μL时,3种目标物均基本实现完全解吸;继续增大解吸体积并不能提高解吸量,反而会影响各目标物色谱峰的分离度和峰形。因此实验选择解吸体积为 500 μL。
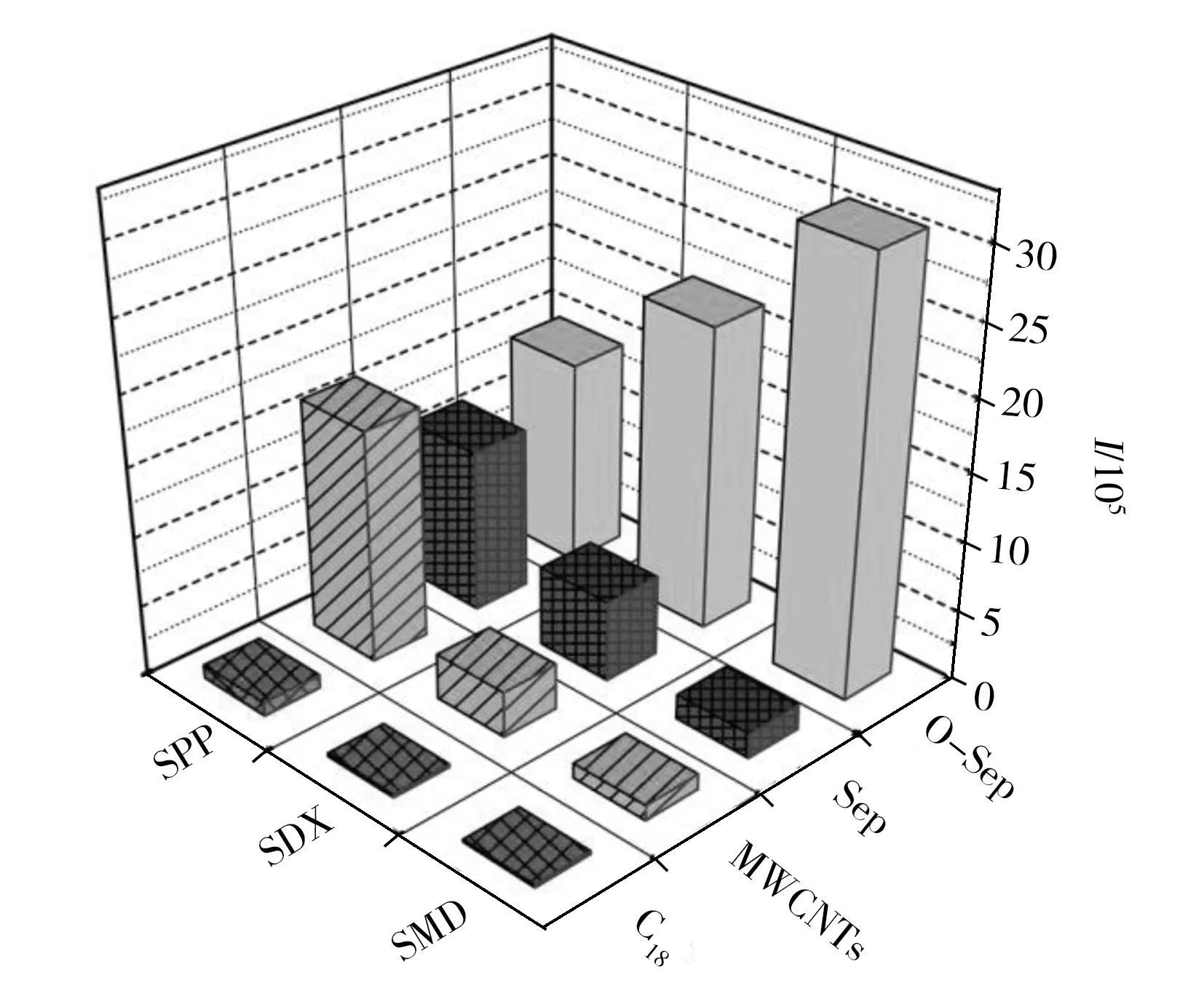
图3 O-Sep与其它材料的萃取性能比较Fig.3 Comparison of extraction performances of O-Sep with other three materials
2.3 吸附容量的研究
分别保持吸附剂用量为10 mg,萃取体积为 3 mL,通过改变萃取溶液质量浓度(由20 μg/L增至560 μg/L)考察了O-Sep微柱的萃取容量。结果表明,随着萃取溶液质量浓度的增加,O-Sep柱的萃取量增大,并在萃取溶液质量浓度为480 μg/L时基本达到萃取平衡。此时,O-Sep萃取柱对SMD、SDX、SPP的饱和萃取容量分别为203.2、150.3、102.1 ng。
2.4 材料萃取性能的对比
对比了O-Sep与Sep、商用C18和多壁碳纳米管(MWCNTs)对3种SAs的萃取性能(图3),O-Sep对SMD的萃取能力高于其他3种萃取材料,富集量约为MWCNTs的 8.5 倍,是C18的 10.2 倍;对SDX的富集量约为MWCNTs的 4.5倍,C18的 8.2倍;然而其对SPP的萃取能力相较于MWCNTs和Sep并无明显提高。这可能是由于O-Sep经酸化处理后,Si—O骨架生成Si—OH基,材料的亲水性有所提高,从而增强了其对极性物质的富集能力。
2.5 分析方法的建立与应用
2.5.1分析方法的建立采用O-Sep萃取不同质量浓度的SMD、SDX和SPP混合标准溶液(1.0~50.0 μg/L),按照“1.7”条件进行HPLC分析,以质量浓度(x,μg/L)为横坐标,对应峰面积(y)为纵坐标绘制工作曲线。由表1可知,SMD、SDX和SPP在1.0~50.0 μg/L范围内具有良好的线性关系,相关系数(r2)大于0.97;检出限(信噪比S/N=3)为0.3~0.5 μg/L;采用10.0 μg/L磺胺类混合标准溶液考察方法的精密度,相对标准偏差(RSD,n=5) 为1.1%~3.2%,表明方法具有良好的灵敏度和精密度。
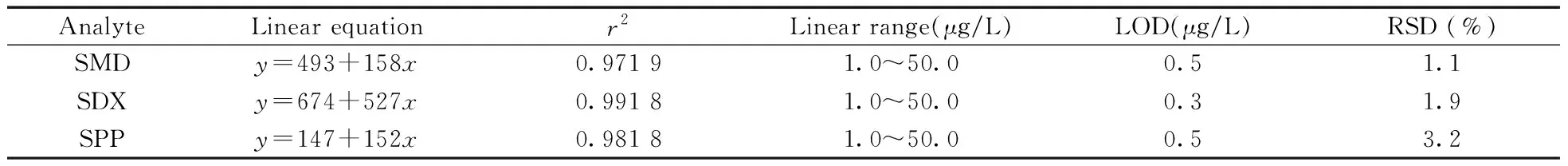
表1 3种目标物的线性方程、相关系数、线性范围、检出限及相对标准偏差Table 1 Linear equations,correlation coefficients,linear ranges,detection limits and RSDs of 3 analytes
2.5.2实际样品的分析将实验室自来水、鱼梁河水和贵阳市4月份雨水按照“1.6”方法处理后,采用O-Sep/SPE/HPLC方法进行测定,均未检出SAs。对上述实际样品进行5.0、10.0、30.0 μg/L 3个质量浓度的加标回收试验,得到3种水样的回收率分别为79.5%~98.3%、74.3%~98.6%和70.2%~98.7%,RSD为1.4%~7.6%(表2)。实际样品加标前后及加标样品在线富集后的色谱图如图4所示,当样品中不加SAs时均未检出SAs;加标10.0 μg/L后,可检测到SAs的色谱峰,但出峰信号较小;而用O-Sep固相萃取柱富集分离后,SAs的色谱峰信号明显升高。结果表明O-Sep固相萃取柱对环境水样中SAs有较好的萃取能力。
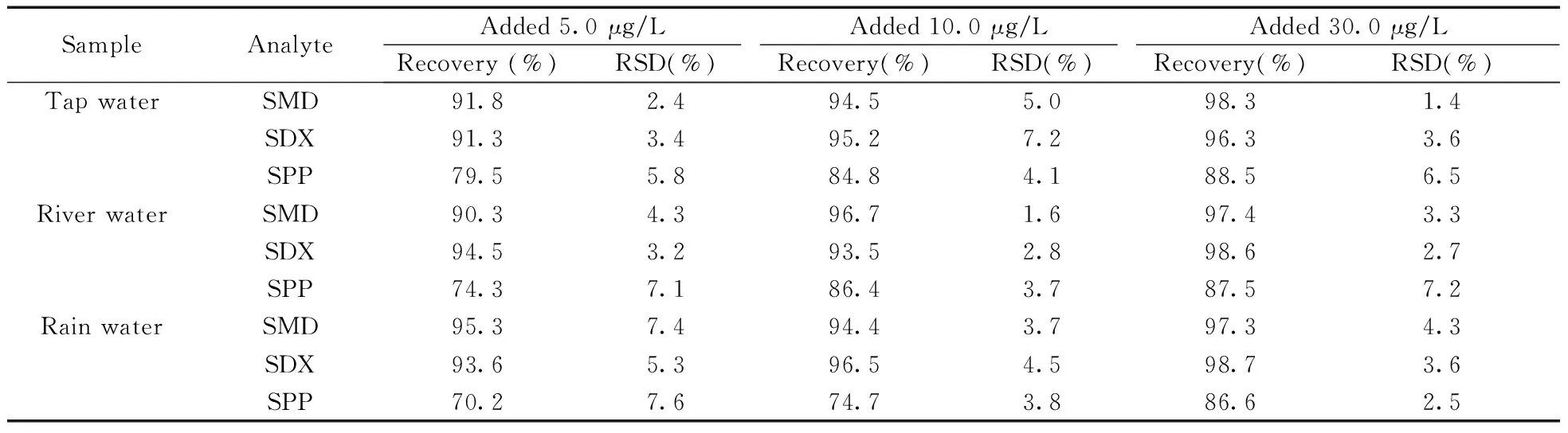
表 2 环境水样中3种磺胺类药物的回收率及相对标准偏差(n=5)Table 2 Recoveries and RSDs of three sulfonamides in spiked environmental water samples(n=5)
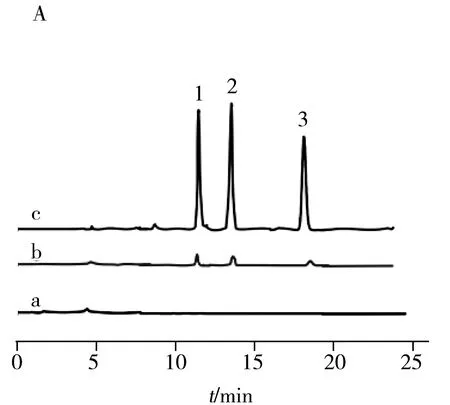
图4 自来水(A)、河水(B)和雨水(C)样品的色谱图
Fig.4 Chromatograms of tap water(A),river water(B) and rain water(C) samples
a:real sample;b:sample spiking with 10.0 μg/L of each compound;c:online enrichment of spiked samples;1.SMD,2.SDX,3.SPP
3 结 论
本文通过酸化法制备了 O-Sep材料,将其填充于自制 PEEK 微柱中,构建了在线萃取装置,优化了萃取解吸条件,研究了O-Sep微柱萃取性能。结果表明填充10 mg O-Sep的微柱对SMD、SDX和SPP的萃取容量分别为203.2、150.3、102.1 ng。所建立的O-Sep/SPE/HPLC在线分析方法实用、简便,在环境水样中磺胺类药物的分析中有较大的应用潜力。
