基于磁球表面的抗原决定基印迹法用于选择性识别细胞色素c的研究
2017-10-19赵晓丽何锡文李文友
赵晓丽, 何锡文, 李文友
(药物化学生物学国家重点实验室,天津市生物传感与分子识别重点实验室,南开大学化学学院,天津 300071)
分子印迹作为一种分离技术,具有预订性、识别性和实用性三大特性。分子印迹聚合物(Molecular Imprinting Polymers,MIPs)具有易于合成、成本低廉、结构稳定等优点,因此被广泛应用在分离提纯[1 - 3]、传感器[4]等领域。目前,对小分子的印迹技术发展已较为成熟,但由于蛋白质分子量大、结构复杂、容易变性等原因,对蛋白质的印迹仍有很多困难[5]。蛋白质的印迹方法主要有包埋法、表面印迹法和抗原决定基印迹法[6 - 7],而研究较多的为表面印迹法和抗原决定基印迹法。抗原决定基印迹法基于抗原-抗体间相互作用的原理,由Rachkov等[8]提出。该方法在印迹过程中只需印迹蛋白质的一小段多肽序列,即该蛋白质的抗原决定基,而非整个蛋白质,得到的印迹聚合物不仅能识别这段多肽序列,还能识别相应的蛋白质。Nishino等[9]分别使用牛血清白蛋白(BSA)、细胞色素c(Cyt c)和乙醇脱氢酶(ADH)C端的九肽作为模板,在进行一系列化学修饰的玻璃表面进行印迹,制得的印迹层对目标蛋白具有很好的吸附选择性。近几年,抗原决定基印迹法已成功地与多种材料结合制备印迹聚合物,如结合电化学方法[10]或使用石英晶体微天平芯片[6,11]的金传感器表面、磁球[12]、量子点[13]等。
Cyt c分子量约11~13 kDa,是生物氧化过程中的电子传递体,从线粒体中泄漏出的Cyt c具有诱导细胞凋亡的作用,因此对Cyt c的分析得到了越来越多的关注[14 - 17]。本工作结合表面印迹法和抗原决定基印迹法,采用一种简便的方法合成了磁性印迹聚合物用于选择性识别Cyt c。使用磁球作为基质,具有制备简单、生物相容性好、低毒、便于快速分离等优点。此外,还利用了多巴胺(DA)自聚的原理,在磁球表面直接形成DA印迹薄膜,洗脱掉模板后得到核壳结构的磁性分子印迹聚合物。合成过程中,磁球表面不再进行其它修饰,直接进行下一步的印迹聚合,操作步骤简单。形成的印迹聚合物采用多种技术手段进行形貌、结构、性质的表征。在吸附实验中,该聚合物的吸附动力学速度很快,对目标蛋白有较好的吸附选择性,并且在多次吸附-洗脱循环中有良好的重复利用性,结果表明使用抗原决定基做模板的聚合物的吸附情况优于使用相应蛋白做模板的情况。
1 实验部分
1.1 仪器和试剂
LC-20AD型高效液相色谱仪(日本,岛津公司);UV-2450型紫外-可见分光光度计(日本,岛津公司);JEM 100CXII型透射电子显微镜(TEM)(日本,电子公司);D/MAX-2500型X-射线衍射仪(XRD)((日本,岛津公司);LDJ 9600-1型振动样品磁强计(VSM)(美国,LDJ电子仪器公司);Vector 22傅立叶变换红外(FT-IR)光谱仪(德国,布鲁克公司)。
Cyt c的C端九肽(AYLKKATNE)购于上海吉尔生化有限公司。FeCl3·6H2O、无水NaAc、乙二醇(EG)、NaH2PO4·2H2O、Na2HPO4·12H2O、十二烷基硫酸钠(SDS)和HAc,均购于天津光复精细化学品厂。甲醇(99.5%)和乙醇(99.7%)购于天津康科德科技有限公司。多巴胺盐酸盐(DA,99%)购于J&K科技(北京)有限公司。所用的蛋白质,包括牛血清白蛋白(BSA,Mw=67 kDa,pI=4.9),Cyt c(Mw=12.4 kDa,pI=9.6)以及辣根过氧化物酶(HRP)(Mw=40 kDa,pI=7.2)购于北京索莱宝科技有限公司。所有试剂均为分析纯或色谱纯。实验用水由艾科浦超纯水系统(重庆颐洋企业发展有限公司)制备。
1.2 Fe3O4磁性纳米粒子的合成
Fe3O4磁性纳米粒子参照文献方法[18],通过溶剂热法合成。将FeCl3·6H2O(1.35 g)和无水NaAc(3.60 g)溶解在EG(35 mL)中,先通过磁力搅拌使其混合均匀,然后将其转移到聚四氟乙烯内衬的不锈钢高压釜中密封,200 ℃反应8 h。反应完成后,待高压釜冷却到室温,通过外加磁场收集得到的产物,分别用乙醇和超纯水洗涤数次,然后用磁铁将产品分离出来,45 ℃下干燥过夜。
1.3 Fe3O4@EMIPs和Fe3O4@CMIPs的合成
称取100 mg Fe3O4纳米粒子分散在10 mL 10 mmol/L的磷酸盐缓冲溶液(PBS,pH=8.0)中,超声30 min使其分散均匀。称取20 mg Cyt c的抗原决定基,并用5 mL上述PBS溶解。然后将二者混合,机械搅拌2 h使其进行充分的预组装。称取60 mg DA溶于5 mL PBS(pH=8.0)中,将溶解好的DA溶液加入上述混合体系中,继续机械搅拌3 h,该过程中保证有充足的空气进入体系,使DA进行聚合反应。反应结束后在外加磁场下分离产物,弃去液体部分,产物用甲醇-HAc(9/1,V/V)的混合液洗脱48 h,除去聚合物中的抗原决定基模板。最后用乙醇和水反复洗涤产物,45 ℃干燥最终的印迹聚合物。非印迹的Fe3O4@ENIPs用同样的方法制备,只是不加入Cyt c的抗原决定基。
Fe3O4@CMIPs(使用Cyt c作为模板)的合成方法同Fe3O4@EMIPs,只是使用20 mg的Cyt c蛋白质作为模板。得到的产物用5%SDS-5%HAc洗脱液洗脱。非印迹的Fe3O4@CNIPs用同样的方法制备,只是不加入 Cyt c。
1.4 多巴胺用量的优化
DA用量不同,形成的聚合物的印迹层厚度不同,因此吸附效果也不同。为使Fe3O4@EMIPs达到最佳的印迹效果,对合成过程中DA的用量进行了优化。固定载体Fe3O4的用量及其它反应条件,DA分别为20、40、60、80、100 mg五种用量,合成了五组不同的Fe3O4@EMIPs和Fe3O4@ENIPs,然后在相同条件下进行吸附性能实验。将5 mg Fe3O4@EMIPs或Fe3O4@ENIPs置于2 mL 0.5 mg/mL的Cyt c的PBS(10 mmol/L,pH=7.0)中,室温下振荡吸附30 min使吸附达到饱和。在外加磁场下分离聚合物和上清液,用紫外-可见分光光度计检测上清液中Cyt c的浓度,进而计算出吸附容量和印迹因子,根据吸附效果选取DA的最佳用量。
1.5 吸附实验
动力学吸附实验:将5 mg Fe3O4@EMIPs或Fe3O4@ENIPs置于2 mL 0.5 mg/mL Cyt c的PBS(10 mmol/L,pH=7.0)中,室温下分别进行2 min到60 min不同时间的振荡吸附。外加磁场下分离聚合物和上清液,用紫外-可见分光光度计检测上清液中Cyt c的浓度。磁性印迹纳米粒子对蛋白质的吸附容量(Q)可以根据如下公式计算:
Q=(c0-ct)·V/m
(1)
其中,Q(mg/g)是吸附容量,c0(mg/g)是蛋白质溶液的起始浓度,ct(mg/g)是吸附后上清液中蛋白质溶液的浓度,V(mL)是蛋白质溶液的体积,m(g)是使用的纳米粒子的质量。
等温吸附实验:将5 mg Fe3O4@EMIPs或Fe3O4@ENIPs置于2 mL不同浓度的Cyt c(0.1 mg/mL到1.0 mg/mL)的PBS(10 mmol/L,pH=7.0)中,室温下分别进行30 min的振荡吸附。然后在外加磁场下分离聚合物和上清液,用紫外-可见分光光度计检测上清液中Cyt c的浓度。同样根据上述公式计算吸附容量。
选择性吸附实验:将5 mg Fe3O4@EMIPs或Fe3O4@ENIPs置于2 mL 0.5 mg/mL的不同蛋白质(Cyt c、BSA、HRP)的PBS(10 mmol/L,pH=7.0)中,室温下分别进行30 min的振荡吸附。外加磁场下分离聚合物和上清液,然后使用高效液相色谱仪检测上清液中不同蛋白质的浓度。印迹聚合物的吸附性能可以通过吸附容量Q和印迹因子(IF)来评估。其中,IF=QMIP/QNIP。QMIP和QNIP(mg/g)分别为印迹聚合物和非印迹聚合物对目标蛋白质的吸附容量。
重复利用性实验:为了检验该印迹聚合物的重复利用性能,5 mg的Fe3O4@EMIPs或Fe3O4@ENIPs分别加入2 mL 0.5 mg/mL Cyt c溶液(10 mmol/L PBS,pH=7.0)中,在室温下振荡吸附30 min直至达到吸附平衡。然后纳米粒子通过外部磁场与溶液分离,用5%SDS -5%HAc的混合溶液洗脱24 h,除去吸附的蛋白质。重新得到的纳米粒子用于下次的吸附-洗脱循环,整个过程重复三次,分别计算每次吸附过程的吸附容量。
1.6 印迹聚合物的表征
用透射电子显微镜(TEM)表征Fe3O4和Fe3O4@EMIPs的形貌特征。用X-射线衍射(XRD)仪表征Fe3O4,Fe3O4@EMIPs和Fe3O4@ENIPs的晶体结构,2θ角在3°~80°范围内。傅立叶变换红外(FT-IR)光谱用Vector 22 FT-IR光谱仪测量。磁性特征用LDJ 9600-1振动样品磁强计(VSM)测量。紫外-可见吸收光谱用UV-2450分光光度计测得。
2 结果与讨论
2.1 核壳型Fe3O4@EMIPs的制备
Fe3O4@EMIPs的制备过程如图1所示。先以溶剂热法合成Fe3O4磁性纳米粒子,然后加入Cyt c的抗原决定基预组装2 h,再加入DA溶液进行聚合反应3 h,从而将模板包覆在磁球表面。待洗脱掉模板分子后,即得到Fe3O4@EMIPs。作为对照,Fe3O4@ENIPs用同样的方法合成,只是不加入模板分子Cyt c的抗原决定基。

图1 Fe3O4@EMIPs的制备示意图Fig.1 Synthesis illustration of Fe3O4@EMIPs
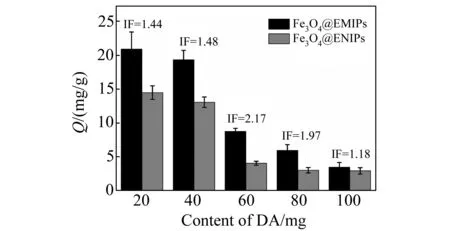
图2 多巴胺的用量对吸附效率的影响Fig.2 Influence of the amount of dopamine on adsorption efficieacy
实验中,我们考察了不同DA用量下聚合物的吸附性能,实验结果如图2所示。根据实验结果,我们可以做如下解释:当DA用量较小(20和40 mg)时,Fe3O4表面没有被完全包裹住,有部分裸露的磁球,因此非特异性吸附较强,Fe3O4@EMIPs和Fe3O4@ENIPs的吸附容量Q均较大,印迹因子IF较小。当DA用量过大(80和100 mg)时,Fe3O4表面聚合的印迹层又过厚,导致模板被包裹在印迹层较深的位置,不易被洗脱出来,可识别的印迹位点变少或是因为印迹位点被包覆在印迹层较深的位置,目标蛋白不易到达印迹位点。这都导致Fe3O4@EMIPs和Fe3O4@ENIPs 的吸附容量Q较小。当DA用量为60 mg时较为合适,聚合物的Q和IF(IF=2.17)都较为理想。所以,在合成此磁性印迹聚合物时,选择DA的用量为60 mg。
2.2 磁性印迹聚合物的表征
采用透射电镜(TEM)观察Fe3O4和Fe3O4@EMIPs的粒径和形貌结构。如图3(a)所示,Fe3O4粒子为球形,分散较为均匀,粒径大小较为统一,约为350 nm。而Fe3O4@EMIPs的粒径(图3(b)和3(c))较Fe3O4相比仅增长约5 nm,可见在Fe3O4表面形成了很薄的一层聚多巴胺(PDA)印迹层。在图3(c)中印迹聚合物的外层可以较为明显地观察到这层PDA。TEM图较为直观地表明了核壳型磁性印迹聚合物的成功合成。Fe3O4表面这层较薄的聚多巴胺印迹层对模板的洗脱及对蛋白质的吸附都非常有利,这使该印迹聚合物有较快的吸附动力学,并且其饱和磁化强度仍能保持较高的数值,便于与溶液达到快速分离。

图3 Fe3O4(a),Fe3O4@EMIPs(b和c)的透射电镜(TEM)图Fig.3 TEM images of Fe3O4(a) and Fe3O4@EMIPs(b and c)
Fe3O4、Fe3O4@EMIPs和Fe3O4@ENIPs的晶型用X-射线衍射(XRD)仪进行分析(图4)。如图所示,当2θ在3°~80°的范围内,三个样品均能明显地观察到Fe3O4的2θ=30.38°、35.58°、43.14°、53.48°、57.08°和62.66° 六个特征峰。与2θ值相对应的峰位置的指数分别为(220)、(311)、(400)、(422)、(511)和(440)。这些峰位置的指数均与JCPDS卡(19-629)磁铁矿的数据相对应。因此,XRD表征说明制得的Fe3O4有很好的晶型,并且在形成聚合物后,其晶型并未发生变化。
Fe3O4和Fe3O4@EMIPs纳米粒子的傅立叶红外(FT-IR)光谱图如图5所示。590 cm-1处为Fe-O的伸缩振动峰,是Fe3O4的特征吸收峰。与裸磁球的光谱图对比,在Fe3O4@EMIPs中,1 640 cm-1处代表芳香环的伸缩振动,表明在磁球表面成功地包覆了PDA层。因此,FT-IR光谱图也可以表明该印迹聚合物的成功合成。

图4 Fe3O4 (a)、Fe3O4@EMIPs(b)和Fe3O4@ENIPs(c)的X-射线衍射(XRD)图Fig.4 XRD patterns of Fe3O4 (a),Fe3O4@EMIPs(b) and Fe3O4@ENIPs(c)
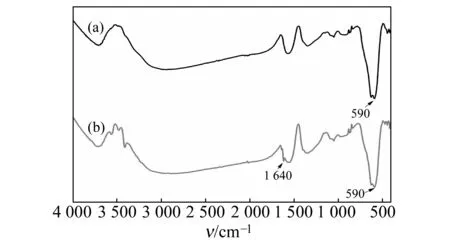
图5 Fe3O4 (a)和Fe3O4@EMIPs(b)的傅立叶红外(FT-IR)光谱图Fig.5 FT-IR spectra of Fe3O4 (a) and Fe3O4@EMIPs(b)

图6 Fe3O4 (a)和Fe3O4@EMIPs(b)的磁滞回线图Fig.6 Hysteresis loops of Fe3O4 (a) and Fe3O4@EMIPs(b)
采用振动样品磁强计(VSM)表征合成的Fe3O4和Fe3O4@EMIPs的磁性质,结果见图6。Fe3O4和Fe3O4@EMIPs的饱和磁化强度分别为60.75、50.96 emu/g。可见,Fe3O4@EMIPs与裸磁球相比,磁化强度略有下降,但仍有较高的磁化强度以保证该印迹聚合物在外加磁场下能和溶液快速分离。此外,饱和磁化强度的下降也侧面说明印迹层的成功形成。
2.3 Fe3O4@EMIPs和Fe3O4@ENIPs的吸附特性
2.3.1吸附动力学称取5 mg Fe3O4@EMIPs和Fe3O4@ENIPs,分别置于2.0 mL初始浓度为0.5 mg/mL 的Cyt c溶液中。Fe3O4@EMIPs 和 Fe3O4@ENIPs 的动力学吸附如图7所示。在0~20 min内,随吸附时间增长,吸附容量逐渐增大。这是因为在吸附的最初阶段,空的印迹位点较多,Cyt c能较容易地进入这些空的印迹位点。而吸附到20 min时吸附达到饱和。其后,随吸附时间的增长,吸附容量不再增大。在吸附过程中,Fe3O4@EMIPs的吸附容量始终高于Fe3O4@ENIPs的吸附容量,由此可以说明在Fe3O4@EMIPs表面成功形成了特异性的印迹位点。从图中可以看出,Fe3O4@EMIPs能在20 min内就达到吸附平衡,主要是因为我们合成的为核壳型分子印迹聚合物,形成的印迹层很薄,位于或接近磁球的表面,因此传质阻力很小,目标蛋白质较容易进入识别位点,使吸附达到平衡。
2.3.2吸附等温线Fe3O4@EMIPs和Fe3O4@ENIPs的等温吸附曲线如图8所示。当Cyt c浓度小于0.5 mg/mL时,随浓度的增大,Fe3O4@EMIPs和Fe3O4@ENIPs的吸附容量逐渐增大。当Cyt c的浓度大于0.5 mg/mL时吸附达到平衡,Fe3O4@EMIPs和Fe3O4@ENIPs的吸附容量不再随蛋白质浓度的增大而增大。在实验考察的浓度范围内,Fe3O4@EMIPs的吸附容量明显大于Fe3O4@ENIPs的吸附容量。当吸附达到平衡时,Fe3O4@EMIPs的饱和吸附容量为8.33 mg/g,印迹因子为2.08。由此可见,在Fe3O4@EMIPs 表面形成了具有特异性识别能力的印迹孔穴,这些印迹孔穴可以吸附目标蛋白质。此外,在Fe3O4@ENIPs表面也有一定的非特异性识别位点,所以非印迹聚合物也存在一定的吸附容量。

图7 Fe3O4@EMIPs和Fe3O4@ENIPs动力学吸附图Fig.7 Adsorption kinetics of Fe3O4@EMIPs and Fe3O4@ENIPs

图8 Fe3O4@EMIPs和Fe3O4@ENIPs等温吸附图Fig.8 Adsorption isotherms of Fe3O4@EMIPs and Fe3O4@ENIPs
对于Fe3O4@EMIPs和Fe3O4@ENIPs的吸附数据,进一步通过Langmuir等温吸附模型来分析。Langmuir方程为:
ce/Qe=1/(QmKL)+ce/Qm
(2)
式中,Qe(mg/g)为Fe3O4@EMIPs或Fe3O4@ENIPs对Cyt c的平衡吸附容量,Qm(mg/g)是理论最大吸附容量,ce(mg/mL)是吸附达到平衡时Cyt c的浓度,KL(mL/mg)是Langmuir吸附平衡常数。
Qm和KL的值可通过Langmuir拟合直线的斜率和截距计算得出,得到的数值列在表1中。线性回归系数(r)高于0.950,说明Langmuir模型比较符合该印迹聚合物和非印迹聚合物。

表1 Fe3O4@EMIPs和Fe3O4@ENIPs的Langmuir模型
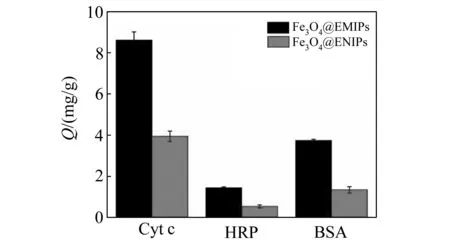
图9 Fe3O4@EMIPs和Fe3O4@ENIPs对三种蛋白的吸附情况Fig.9 Binding specificity of Fe3O4@EMIPs and Fe3O4@ENIPs toward three kinds of proteins
2.3.3吸附选择性为了考察合成的Fe3O4@EMIPs的特异性吸附能力,我们采用了两种与Cyt c分子量和等电点不同的蛋白质作为对照进行吸附实验。达到吸附平衡后,通过外加磁场分离上清液和纳米粒子,用高效液相色谱法检测上清液中剩余蛋白质的含量,所得结果如图9所示。从图中可以看出,该印迹聚合物对目标蛋白有较高的吸附容量。对于HRP和BSA两种蛋白,其吸附容量均较小。
2.3.4重复利用性为了考察合成的Fe3O4@EMIPs和Fe3O4@ENIPs的重复利用性,用一定量Fe3O4@EMIPs和Fe3O4@ENIPs进行了三次吸附-洗脱循环实验(图10)。在洗脱过程中,我们使用5%SDS-5%HAc洗脱吸附的 Cyt c。在实验中,发现用SDS-HAc洗脱液洗脱蛋白质的效果优于用甲醇-HAc的洗脱效果,因此此处改用SDS-HAc洗脱液洗脱吸附的蛋白质。从图10可以看出,在吸附-洗脱循环中,Fe3O4@EMIPs和Fe3O4@ENIPs的吸附容量都略有下降,可能是因为一些印迹位点在洗脱过程中被破坏,或是因为一些印迹位点吸附的蛋白质较难被洗脱下来,导致这些印迹位点在下次吸附实验中不能再吸附蛋白质。但是Fe3O4@EMIPs和Fe3O4@ENIPs的吸附容量下降均较小,吸附容量仍维持在原来的89.0%以上。此外,印迹因子在三次吸附-洗脱循环中变化不大,均维持在2.0左右。由此可以说明,该聚合物具有良好的可重复利用性。
2.4 Fe3O4@EMIPs和Fe3O4@CMIPs吸附性能的比较
为了说明抗原决定基印迹法的优越性,我们分别以Cyt c的抗原决定基和Cyt c为模板合成了两种印迹聚合物Fe3O4@EMIPs和Fe3O4@CMIPs,然后分别称取5 mg聚合物振荡吸附0.5 mg/mL的Cyt c溶液,直至达到饱和。结果(图11)表明,Fe3O4@EMIPs的吸附容量(8.47 mg/g)大于Fe3O4@CMIPs的吸附量(4.85 mg/g),印迹因子也明显优于蛋白质做模板的情况。
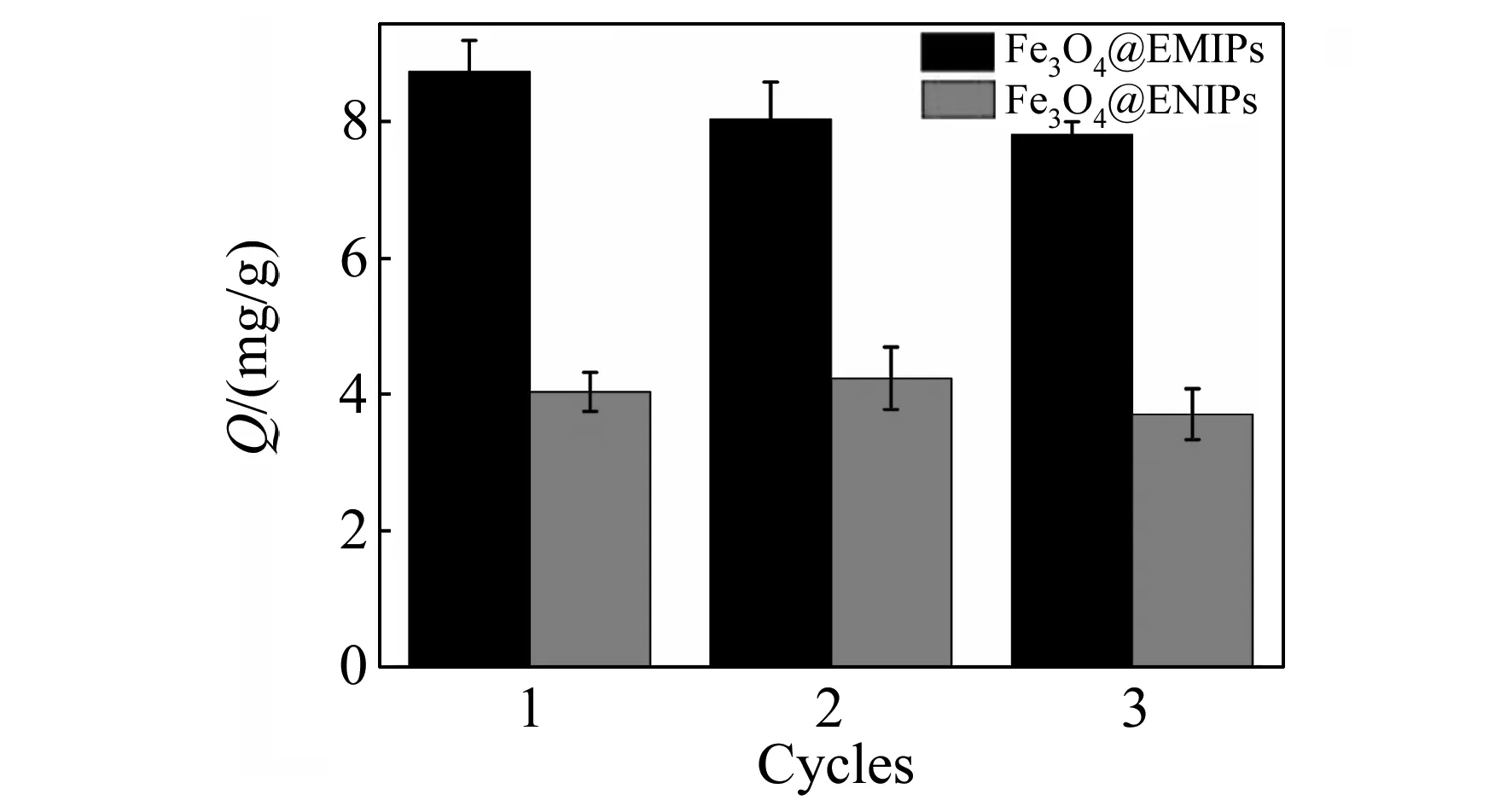
图10 Fe3O4@EMIPs和Fe3O4@ENIPs的重复利用性实验Fig.10 Reusability of Fe3O4@EMIPs and Fe3O4-@ENIPs
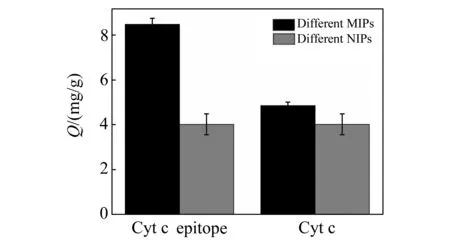
图11 Fe3O4@EMIPs和Fe3O4@CMIPs对Cyt c的吸附情况Fig.11 Binding amount of Fe3O4@EMIPs and Fe3O4-@CMIPs towards Cyt c
3 结论
本工作结合表面印迹法和抗原决定基印迹法合成了核壳型的磁性分子印迹聚合物,并利用了DA在弱碱性条件下自聚的特点,在磁球表面直接进行聚合。该方法操作简单、制备过程中无需加热。形成的印迹聚合物不仅有磁性纳米粒子易于分离的优点,又有表面印迹吸附动力学快的优点。此外,该印迹聚合物对目标蛋白有较好的吸附选择性,且在三次吸附-洗脱循环中吸附容量略有下降,具有良好的可重复利用性。该方法为制备抗原决定基印迹聚合物提供了一种简便快速的途径,并且丰富了抗原决定基印迹法的研究。
