3例杜氏肌营养不良家系基因诊断策略探讨*
2016-09-21陈远春
陈远春,代 英,钟 敏
(1.重庆医科大学附属儿童医院神经内科 400014; 2.重庆涪陵中心医院儿科 408000;3.重庆医科大学附属儿童医院儿童保健科 400014; 4.儿科学重庆市重点实验室/儿童发育疾病研究教育部重点实验室 400014)
·技术与方法·
3例杜氏肌营养不良家系基因诊断策略探讨*
陈远春1,2,代英3,4,钟敏1.4△
(1.重庆医科大学附属儿童医院神经内科400014; 2.重庆涪陵中心医院儿科408000;3.重庆医科大学附属儿童医院儿童保健科400014; 4.儿科学重庆市重点实验室/儿童发育疾病研究教育部重点实验室400014)
目的对3例既往多重PCR基因检测阴性的杜氏肌营养不良(DMD)家系进一步进行基因诊断及家系成员遗传指导。方法收集先证者及其家系成员的临床资料和基因组DNA,多重连接依赖的探针扩增(MLPA)或第2代高通量测序对DNA样本进行DMD基因突变检测。结果家系1检测到3名男性Exon 7缺失,2名女性杂合子携带者。家系2先证者chrX-32486626的Exon 23发现c.3127C>T,其母chrX-32486626存在c.3127C>T杂合突变,患儿母亲目前孕中胎儿系女孩。家系3先证者chrX-32509581的Exon 20发现c.2411G>A,其母chrX-32509581存在c.2411G>A的杂合突变。结论MLPA或联合第2代高通量测序能够有效基因确诊DMD患者及其家系成员,为遗传咨询及产前诊断提供依据。
肌营养不良,杜氏;基因;突变;高通量测序
杜氏肌营养不良(duchenne muscular dystrophy,DMD) 是由位于Xp21.2的DMD基因缺陷所致的X连锁隐性遗传病,活产男婴发病率约1/(3 600~6 000)[1]。该编码基因包含79个外显子、78个内含子和8个启动子,已报道的基因突变多达7 000余种,突变类型多样:大片段缺失突变约60%,大片段重复占7%,微小缺失或插入占7%,单碱基点突变占20%,剪切位点突变占2%,还包括少量复杂突变和内含子突变[2-5]。本研究中3例DMD家系既往多重PCR基因检测均阴性,通过多重连接探针扩增技术(MLPA)和第2代高通量测序最终找到致病的基因突变位点而确诊,为家系成员遗传咨询及产前诊断提供了准确依据。
1 资料与方法
1.1一般资料家系1,先证者1,男,6岁半,G7P5,否认产伤窒息史,1岁3个月会走路。自4岁开始渐渐出现肢体乏力,行走缓慢,鸭步,易跌跤,渐加重至蹲起困难,双小腿增粗变硬。查体:营养中等,鸭步,Gower征(+),双侧腓肠肌中度肥大,触之稍硬,双下肢肌力Ⅳ级。肌电图示肌源性损害;血肌酸激酶4 050 U/L(参考值50~220 U/L)。重庆医科大学附属儿童医院多重PCR检测DMD基因热点区域基因缺失未见异常。家系调查:患儿父母体健,非近亲结婚,患儿大哥幼年有类似病史,现在19+岁,已经瘫卧于床,全身肌肉萎缩。Ⅲ4(第3代第4名家系成员)现年3+岁,腓肠肌轻度肥大,肌力、肌张力尚正常。家系2,先证者2,男,7岁,2013年重庆医科大学附属儿童医院临床诊断为DMD,因患儿母亲再次怀孕6个月,为评估此次怀孕胎儿患病风险而来院复诊。G1P1,无产伤窒息史。1岁半走路。近4岁开始渐渐出现行走乏力,渐至蹲起困难,鸭步。查体:营养尚可,心音有力,无心界扩大,鸭步步态,Gower征(+),双侧腓肠肌中度肥大,触之稍硬,双下肢肌力Ⅳ级。肌电图示肌源性损害;血肌酸激酶12 400 U/L;2013年重庆医科大学附属儿童医院多重PCR基因检测未见异常。家族史无异。家系3,先证者3,男,5岁,2015年初重庆医科大学附属儿童医院临床诊断DMD,为进一步了解再次生育后代患病风险而来院就诊。患儿系G1P1,无产伤窒息史。1岁2个月走路。4岁半渐出现行走易累,上台阶乏力。查体:营养尚可,未见鸭步步态,Gower征(±),双侧腓肠肌轻度肥大,单足跳稍差。肌电图示肌源性损害;血肌酸激酶5 100 U/L;2015年初重庆医科大学附属儿童医院多重PCR基因检测未见异常。家族史无异。
1.2方法
1.2.1基因组DNA的提取患儿监护人签署知情同意书,所有研究经重庆医科大学附属儿童医院临床医学伦理研究委员会同意。取患儿及其父母亲外周血各2 mL,家系1加取其他4位同胞兄弟姐妹、外祖父母及祖父母外周血各2 mL,乙二胺四乙酸钠抗凝。用基因组DNA提取试剂盒(QIAamp DNA Blood Midi Kit,Qiagen,Hilden,Germany)提取DNA。
1.2.2MLPA购买荷兰MRC-Holland公司检测试剂盒SALSAP034和P035。根据说明书操作,基因组DNA变性后与SALSA MLPA探针杂交,杂交时间16 h,加入连接缓冲液和连接酶进行连接反应,经PCR扩增后用ABI 3130基因分析仪进行毛细管电泳,所得数据用Gene Marker v1.6软件分析,通过计算健康对照人群平均扩增峰值,对所扩增峰值进行标准化处理,分析得出MLPA分析结果图。每次MLPA检测都包括一个健康对照样本,检测结果重复2次。发现致病的基因突变即确诊,未发现异常的继续进行第2代高通量的测序。
1.2.3第2代高通量测序利用Covaris S220超声波仪(massachusetts,USA)将先证者2和3基因组DNA打断成200~250 bp的片段。将1 μg提纯的DNA片段进行末端修复并连接上标记性接头,完成单个患者的DNA建库。用定制的基因片段捕获芯片SureSelect DNA Target Enrichment Baits (Agilent Technologies,Santa Clara,USA),与2~4个标记好的患者DNA库杂交16 h,除去未结合的片段。洗脱芯片特异性筛选的片段,再进行杂交后扩增,最后进行高通量测序仪Illumina HiSeq2000 Analyzers (Illumina,San Diego,USA)连续测序200个循环,并用Illumina bcl2fastq Conversion (Version 1.8.4)读出原始测序数据。用此前经过验证的过滤原则对原始数据进行筛选,生成双端100 bp的过滤后数据。再将这些数据利用软件(Burrows Wheeler Aligner Multi.Visionsoftware package)与参考序列[the reference human genome from the NCBI database(Build 37)]进行比对,输出测序结果。最后利用SOAPsnp软件和Samtools pileup软件找出点突变、微小缺失和插入。
1.2.4Sanger法验证对于先证者2和3发现的致病突变,在其所在片段上下游设计引物,PCR扩增并对产物做Sanger测序,所得结果与DMD基因标准序列(NG_012232.1)进行比对,从而验证基因芯片捕获和高通量测序的结果。两位先证者的父母也进行该法验证。
2 结 果
家系1的家系图如图1所示,先证者1的MLPA检测发现DMD基因Exon 7缺失。Ⅲ1和Ⅲ4均发现DMD基因Exon 7缺失,结合临床均确诊为DMD。Ⅰ4和Ⅱ2均系该Exon 7基因缺失杂合子携带者。
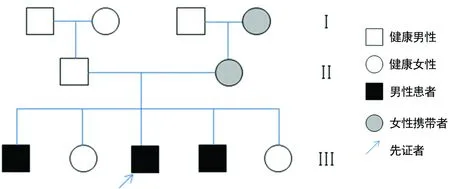
图1 先证者1家系图
家系2和3 MLPA检测未见致病的基因突变,故进行第2代高通量测序。先证者2的检测结果显示:chrX-32486626位置Exon 23区域发现一处半合子突变:c.3127C>T。家系验证结果:其母chrX-32486626存在c.3127C>T的杂合突变,其父未见异常,见图2~3。患儿母亲到产科进行医学必要胎儿性别鉴定系女孩。先证者3结果:chrX-32509581位置Exon 20区域发现一处半合子突变:c.2411G>A。家系验证结果:其母chrX-32509581存在c.2411G>A的杂合突变,其父未见异常。查询HGMDpro数据库发现先证者2和3的突变系致病突变。
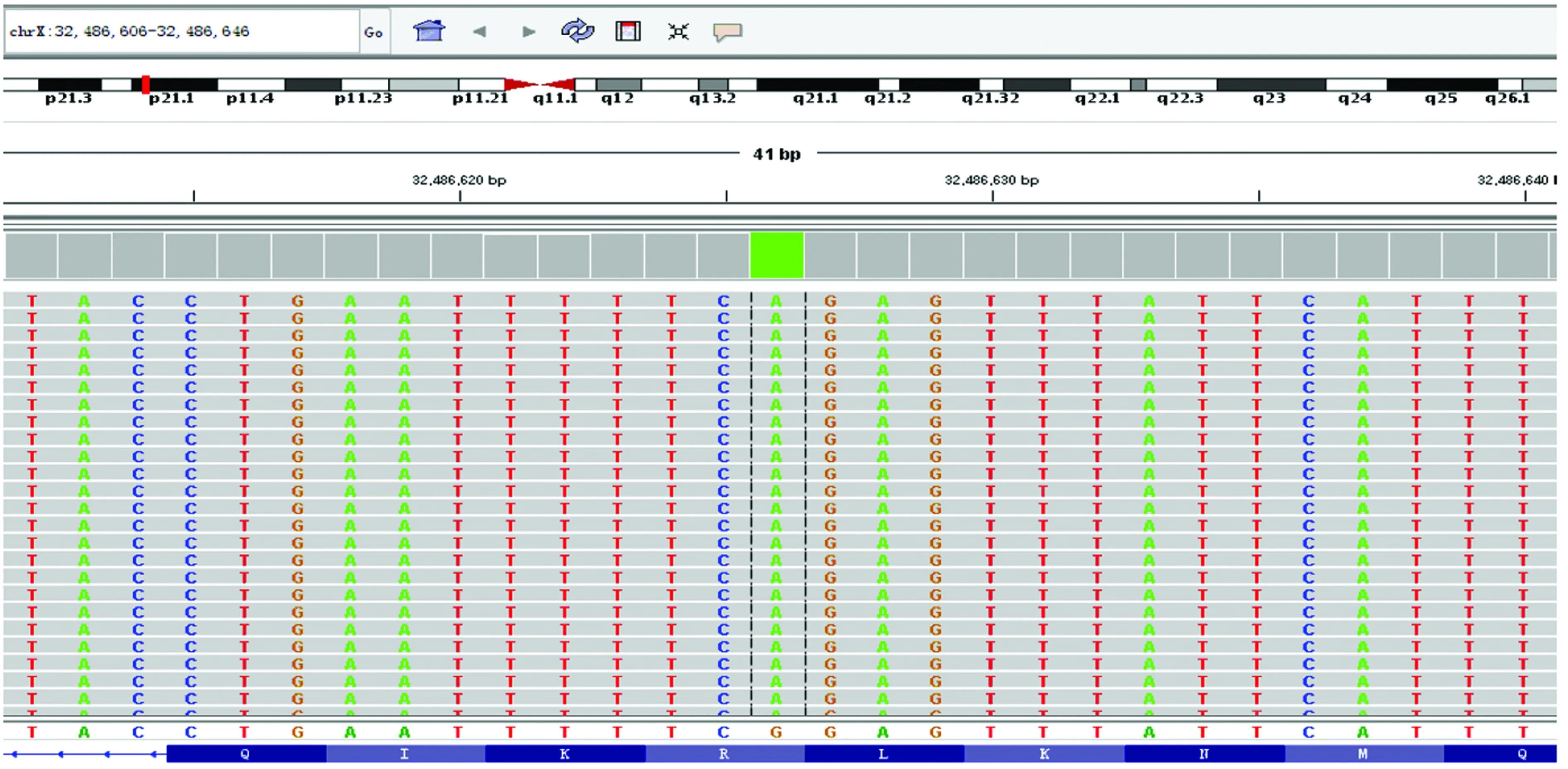
突变位置用黑色虚线框出。
图2患儿DMD基因chrX-32486626区域Exon 23:c.3127C>T
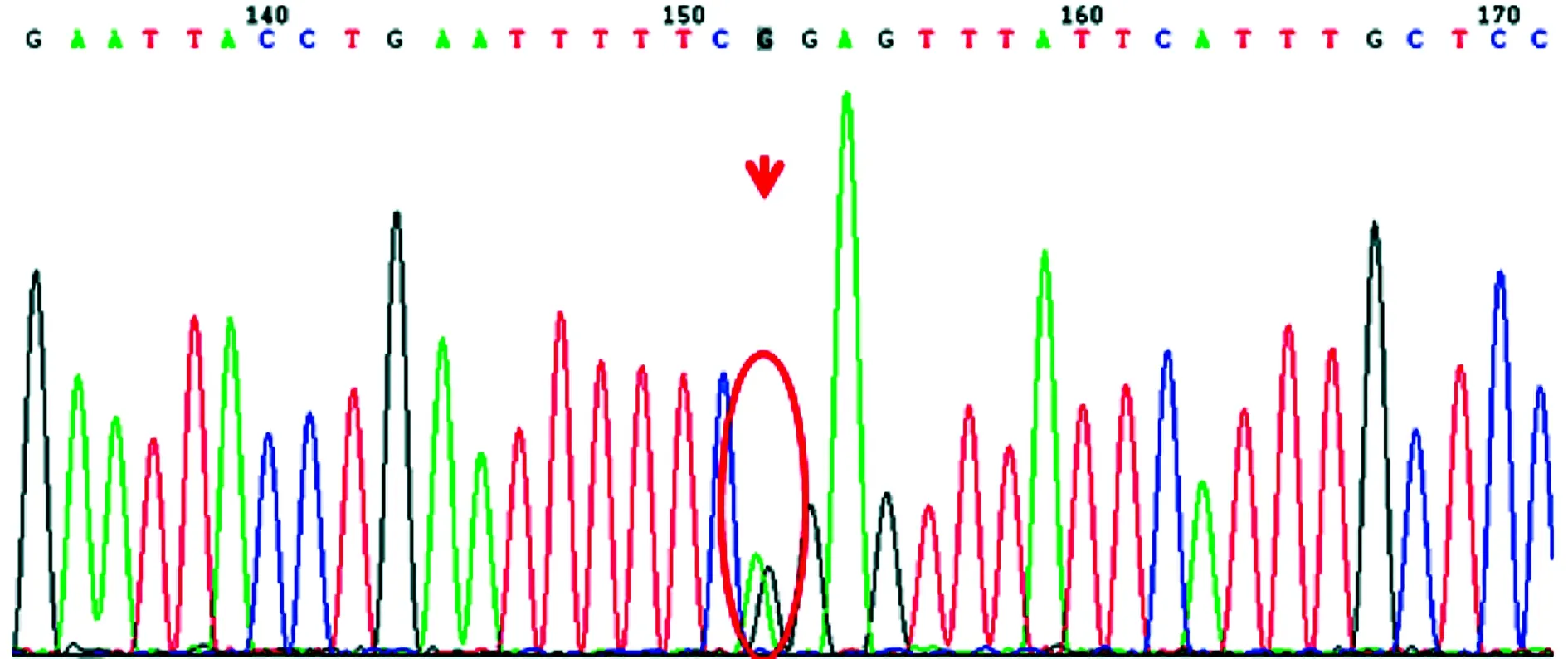
突变位置用红色实线及箭头标出。
图3患儿母chrX-32486626存在c.3127C>T的杂合
突变
3 讨 论
DMD是儿科常见的遗传性神经肌肉病,发病男性占绝大多数,女性常为携带者,约2/3 患儿的病变基因来自于母亲,近1/3 病例系新发突变[6]。一般3~5岁发病,全身乏力呈进行性加重,逐渐卧床,最终因心功能不全和(或)呼吸衰竭多于20岁左右死亡,严重影响儿童的身心健康[1-2]。近年来,随着口服激素等治疗方案的发展,患者的生存期和生活质量较以往有了一定改善。但是,本病迄今为止仍缺乏有效的根治方法。因此,高效、准确的基因诊断、携带者检测及遗传咨询对有效减少新发病例至关重要[1-2,5]。
多重PCR是DMD的主要基因检测手段,可诊断约30%~40%患者数的DMD基因外显子热点区域大片段缺失,但对于非热区的外显子缺失、非缺失突变和女性携带者不能检测[7-12]。本研究的3个家系多重PCR检测未发现致病的基因突变位点,故进行进一步的基因分析。MLPA能有效检测大片段缺失或重复突变,使基因确诊率提高到70%左右[7-14]。家系1通过该技术确诊包括先证者在内的3名男孩均系DMD患者,同胞的2名姐妹均未携带致病基因,患儿母亲及外婆系突变基因携带者,为患儿家系成员的诊断及遗传咨询提供了有力证据。但是,家系2和3仍未检测到致病的基因突变位点。据报道,通过多重PCR结合MLPA检测,仍有占全部DMD致病突变30%左右的微小突变不能检测到,近年来第2代高通量测序使得这部分微小突变检测成为可能[7-12]。故家系2和3继续进行该2代测序,均发现先证者和携带者的致病基因突变位点。家系2的母亲此次怀孕系女婴,故可正常分娩。遗传指导家系成员的生育风险,最大程度杜绝后代中本病的发生。家系3的先证者母亲被证实系突变致病基因携带者,为后期的再次怀孕给产科提供了准确的依据。本研究的3个家系诊断过程值得临床医生高度关注。
因此,掌握本病正确的基因诊断策略对每个医务工作者都至关重要。从经济角度考虑,对临床表现和普通血生化检测疑诊DMD患者,先行多重PCR分析,阴性者再予MLPA,仍阴性但高度疑诊患者,考虑第2代高通量测序。在检测成本进一步降低后,有望通过该单一检测就能涵盖几乎全部的DMD突变类型及其他上百种肌病的基因突变,缩短了诊断周期,为遗传咨询以及进行产前诊断提供依据,使患者及其家系成员最大程度获益。这在目前DMD尚缺乏有效根治手段的情况下意义尤其重大。
[1]Bushby K,Finkel R,Birnkrant DJ,et al.Diagnosis and management of Duchenne muscular dystrophy,part 1:diagnosis,and pharmacological and psychosocial management[J].Lancet Neurol,2010,9(1):77-93.
[2]Muntoni F,Torelli S,Ferlini A.Dystrophin and mutations:one gene,several proteins,multiple phenotypes[J].Lancet Neurol,2003,2(12):731-740.
[3]Aartsma-Rus A,Van Deutekom JC,Fokkema IF,et al.Entries in the leiden duchenne muscular dystrophy mutation database:an overview of mutation types and paradoxical cases that confirm the reading-frame rule[J].Muscle Nerve,2006,34(2):135-144.
[4]Baskin B,Gibson WT,Ray PN.Duchenne muscular dystrophy caused by a complex rearrangement between intron 43 of the DMD gene and chromosome 4[J].Neuromuscular Disorders,2011,21(3):178-182.
[5]Bladen CL,Salgado D,Monges S,et al.The TREAT-NMD DMD Global database:analysis of more than 7000 duchenne muscular dystrophy mutations[J].Hum Mutat,2015,36(4):395.
[6]Prior TW,Bridgeman SJ.Experience and strategy for the molecular testing of duchenne muscular dystrophy[J].J Mol Diagn,2005,7(3):317-326.
[7]刘敏娟,谢敏,毛君,等.第2代测序技术在假肥大型肌营养不良基因诊断中的应用[J].中华医学遗传学杂志,2012,29(3):249-254.
[8]王晶,贺勇.杜氏肌营养不良分子诊断技术的研究进展[J].国际检验医学杂志,2014,35(10):1307-1309.
[9]戴毅,姚凤霞,魏晓明,等.基因芯片捕获及高通量测序在迪谢内型肌营养不良基因诊断中的初步研究[J].中华神经科杂志,2013,46(3):188-192.
[10]林齐防,陈万金,王柠,等.应用多重连接依赖性探针扩增定量技术检测假肥大型肌营养不良重复突变及携带状态[J].中华神经科杂志,2011,44(8):568-573.
[11]Gintjee TJ,Magh AS,Bertoni C.High throughput screening in duchenne muscular dystrophy:from drug discovery to functional genomics[J].Biology (Basel),2014,3(4):752-780.
[12]杜文津,万琪,陈晋文,等.假肥大型肌营养不良基因突变检测方法的应用比较[J].卒中与神经疾病,2010,17(5):289-292.
[13]WillisAS,van den Veyver I,Eng CM.Multiplex ligation-dependent probe amplification(MLPA)and prenatal diagnosis[J].Prenat Diagn,2012,32(4):315-320.
[14]Zimowski JG,Massalska D,Holding MA,et al.MLPA based detection of mutations in the dystrophin gene of 180 Polish families with Duchenne/Becker muscular dystrophy[J].Neurol Neurochir Pol,2014,48(6):416-422.
Gene diagnosis in 3 family members of Duchenne muscular dystrophy
ChenYuanchun1,2,DaiYing3,4,ZhongMin1,4△
(1.DepartmentofNeurology,Children′sHospitalofChongqingMedicalUniversity,Chongqing400014,China;2.DepartmentofPediatrics,theCentralHospitalofFulin,Chongqing408000,China;3.DepartmentofChildHealth,Children′sHospitalofChongqingMedicalUniversity,Chongqing400014,China;4.ChongqingKeyLaboratoryofPediatrics/MinistryofEducationKeyLaboratoryofChildDevelopmentandDisorders,Chongqing400014,China)
ObjectiveTo perform gene diagnosis of Duchenne muscular dystrophy(DMD) in 3 family members who were negative for DMD gene detected by multiplex PCR and to provide genetic counseling for their family members accordingly.MethodsThe clinical data and genomic DNA of patients and their family members were collected,DMD gene mutation were detected by multiplex ligation dependent probe amplification (MLPA) or the 2nd generation of high-throughput sequencing.ResultsIn the first family,3 male patients were detected deletion of Exon 7 and 2 female were heterozygous carriers.In the second family,it was found in the proband that point mutation of c.3127C>T in the Exon 23 of chrX-32486626 and c.3127C>T heterozygous mutations was confirmed in his mother,the mother was pregnant with a girl.In the third family,point mutation of c.2411G>A was detected in the Exon 20 of chrX-32509581 in the proband and his mother had c.2411G>A heterozygous mutation.ConclusionMLPA or combining with the 2nd generation of high-throughput sequencing can offer effective gene diagnosis for the patients of DMD and their family members,and provide the basis for genetic counseling and prenatal diagnosis.
muscular dystrophy,Duchenne;gene;mutation;high throughput sequencing
陈远春(1978-),主治医师,硕士,主要从事儿科临床诊治工作。△
,E-mail:cy_zm11@126.com。
10.3969/j.issn.1671-8348.2016.07.020
R746.2
A
1671-8348(2016)07-0926-03
2015-09-08
2015-11-20)
