HONO来源及其对空气质量影响研究进展
2014-04-26安俊岭汤宇佳中国科学院大气物理研究所大气边界层物理与大气化学国家重点实验室北京100029
安俊岭,李 颖,汤宇佳,陈 勇,屈 玉 (中国科学院大气物理研究所,大气边界层物理与大气化学国家重点实验室,北京 100029)
HONO来源及其对空气质量影响研究进展
安俊岭*,李 颖,汤宇佳,陈 勇,屈 玉 (中国科学院大气物理研究所,大气边界层物理与大气化学国家重点实验室,北京 100029)
综述了HONO来源(源排放、均相反应和非均相反应生成)、HONO模拟研究以及HONO来源对空气质量的影响.指出均相反应中激发态 NO2与水汽作用形成 HONO的机制在高 NOx排放地区具有重要作用,但反应速率需进一步证实.非均相反应中水解反应可能是HONO最主要来源,空气质量模式模拟结果也支持该观点;soot表面的光照催化反应在soot高排放地区对HONO贡献较大,但仍需大量外场实验证实;土壤排放机理的外场实验研究极少,亟待加强.
气态亚硝酸;OH· ;氮氧化物;气溶胶;非均相反应
OH⋅是大气中最重要的氧化剂,对流层中大多数痕量气体主要与 OH⋅反应而被转化或去除,因此,OH⋅决定着大多数痕量气体在大气中的寿命[1-3].OH⋅浓度水平可作为大气氧化能力的指标,也是局地大气对痕量气体自清洁能力的量度.大气中 OH⋅主要来源有 O3的光解,HONO的光解[2,4]、HCHO的光解[2]、H2O2的光解以及 O3与烯烃的反应[5].
HONO即气态亚硝酸,是城市污染的一种典型代表物,一般在污染严重的城市地区浓度较高.OH⋅主要来源于 HONO光解[6-7].近年来大量研究结果表明,HONO对OH⋅的贡献不仅在清晨,甚至在全天都占有重要地位[6,8-13].据报道,白天HONO光解对OH⋅的贡献可达60%[13].
1979年Perner和Platt利用长光程差分吸收光谱仪在大气中首次观测到HONO[14],此后在该方面开展了不少外场实验研究[4,6-10,13-28].结果普遍表明,HONO在城市等重污染地区浓度较高,在乡村等清洁地区浓度较低(表 1).在日变化方面,HONO通常在夜间不断累积,白天光解作用使其浓度在中午或午后达到最低(量级为10-12V/V).近几年的观测结果表明,HONO白天浓度也可达10-9V/V[11,28],如,智利圣地亚哥春季 HONO白天平均浓度可达1.90×10-9V/V[10],广州HONO白天浓度约2.0×10-9V/V[20].如此高的HONO观测浓度无法用众所周知的气相化学反应生成来解释(1.2节),于是HONO来源及其对空气质量的影响 已成为目前大气化学/大气环境的研究热点.
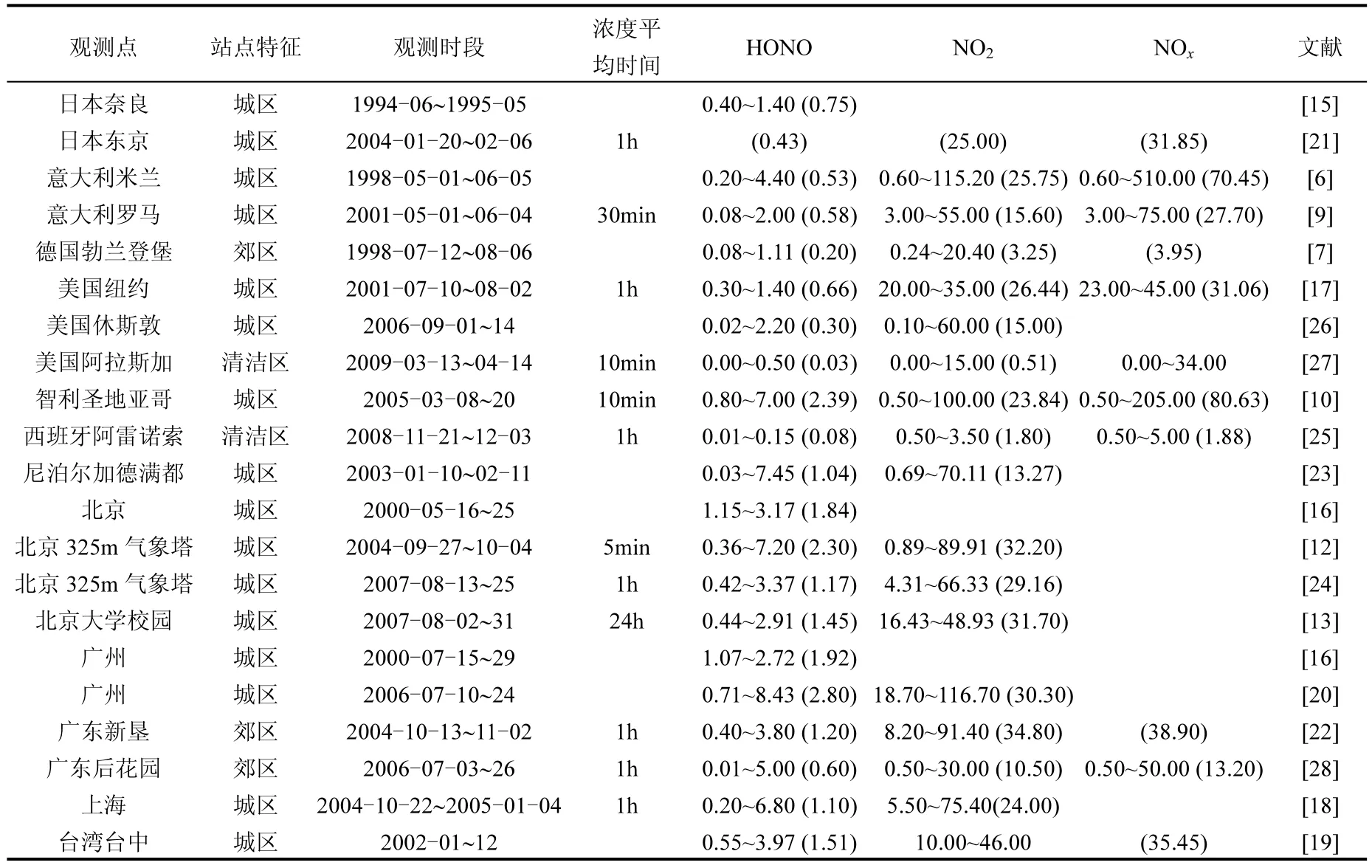
表1 各站点HONO、NO2和NOχ观测值(×10-9V/V)Table 1 Measurements of HONO, NO2and NOχat the studied sites (×10-9V/V)
1 大气中HONO来源
1.1 HONO源排放
1.1.1 燃烧过程排放 燃烧过程排放HONO通常根据机动车尾气排放的HONO占NOχ的比例来量化. 早期经常针对不同车型进行研究,如柴油车排放的HONO约占NOχ排放量的1%,汽油车尾气排放的HONO不足NOχ排放量的0.01% (燃料充足时)和0.15%(燃料匮乏时)[29].由于排放量依赖于发动机的类型和运行状况,现在常利用隧道实验中HONO、NOχ和其他相关参数测量值来估算HONO的排放量,这类实验HONO/NOχ比值的变化范围为 0.3%[30]~0.8%[31]. 0.8%该比值是城市交通高密度区上班期间的典型值,与重污染大气中监测的比例一致,被许多学者采纳[31-34].
1.1.2 土壤排放 Su等[35]指出土壤中的微生物能够通过硝化和反硝化作用将含氮养分转化为与 H+结合生成亚硝酸(R1),亚硝酸通过地气交换过程以气态形式释放到大气中.反应速率与土壤中亚硝酸盐含量、pH值、含水量以及温度有关.该机制不但会影响大气光化学以及全球陆地生态系统,还会影响碳氮循环和气候变化[36].不过,土壤中亚硝酸盐浓度通常都很低[37],相关的外场观测实验极少[38].
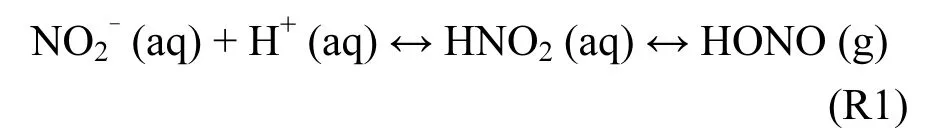
1.2 HONO均相反应生成
均相反应是在单一相态物质中发生的化学反应.HONO最主要的均相反应是HONO光解反应的逆反应R2.该反应在白天OH⋅和NO浓度较高时重要,夜间OH⋅浓度一般较低,该反应贡献较小.某些地区(如珠江三角洲)夜间 OH⋅浓度较高
[39]时,反应R2对夜间HONO的贡献不能不考虑.无论如何,反应R2无法解释白天HONO高观测浓度.Alicke等[6]指出反应R2在中午时段生成HONO 仅几×10-12V/V;Su等[22]发现光化学稳态平衡法计算的HONO浓度比观测值低一个数量级.另外,NO参与的反应(如, NO + NO2+ H2O →2HONO)速率很低,在实际大气中可以忽略.
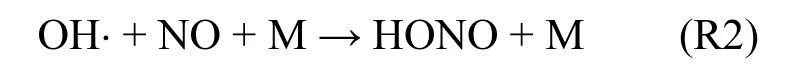
实验室研究发现邻硝基酚光解可产生HONO[40].尽管1.0×10-9V/V硝基酚经太阳光直射可生成HONO达100.0×10-12V/V h-1,但相关机理仍需外场实验验证.根据理论计算,Zhang等[41]提出气态H2O、NO2和NH3经均相核化形成HONO的机制(R3),但反应 R3既未在实验室证实,也未在外场实验观测到.
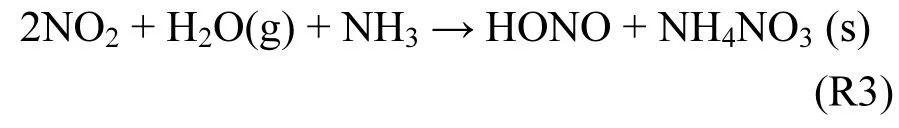
Li等[42]发现由太阳光激发的与空气中水汽反应可产生 HONO(以下简称 NO2*机制),
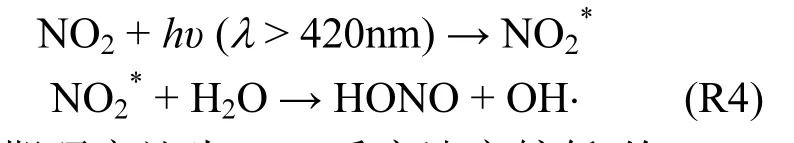
早期研究认为 R4反应速率较低,约 1.2× 10-14cm3/(molecule · s),对HONO和OH⋅的贡献非常有限[43],所以目前大多数空气质量模式并不包含该机制.但Li等[42]认为该反应速率可达1.7× 10-13cm3/(molecule · s),比Crowley等[43]结果高一个数量级,该反应在NOχ排放量较大的地区可能具有非常重要的作用.
1.3 HONO非均相反应生成
发生在大气中颗粒物表面、含表面水层的颗粒物表面、云滴表面的化学转化和光化学过程统称为非均相反应[3].HONO的非均相来源大体可分为3类:水解反应、还原反应和光照催化反应. 1.3.1 水解反应 NO2在各种湿润表面发生的非均相反应 R5可能是 HONO最主要来源
[26,28,44-46],该反应为一级反应[2].R5可能发生在地表面(包括裸露的土壤、城市建筑物表面、雪地等),也可发生在气溶胶表面(包括云滴、雾滴、空气中的颗粒物等).Finlayson-Pitts等[44]通过实验室研究给出了反应 R5在地表面的形成机理,指出由NO2生成的N2O4是驱动该反应发生的重要物种,产物中HONO有一部分脱离地表面返回大气,HNO3则留在反应表面,该反应速率与界面吸附的液态水含量有关.Wojtal等[47]认为反应R5可在海洋表面发生.外场实验表明大气中气溶胶与HONO浓度,或气溶胶比表面积与HONO浓度具有很好的相关性[12,20,48],隐含说明气溶胶表面是 R5发生反应的最主要界面,固定界面(土壤、建筑表面等)可能不重要[38,48].

气溶胶表面生成的 HNO3可重新参与一系列大气化学反应,使大气中硝酸盐浓度增升,气溶胶可通过长距离输送而影响区域或全球对流层化学过程.城市地区大气气溶胶组分非常复杂,可作为云雾形成过程中的凝结核.大量观测表明,在气溶胶和云滴表面可形成 HONO[49].研究表明,京津冀地区气溶胶表面的非均相反应对大气氧化性、霾和能见度可能具有重要影响[12,38,50-51].
1.3.2 还原反应 Ammann等[52]首次提出NO2在烟灰(soot)表面可生成HONO(R6).由于soot反应后几分钟内迅速丧失活性,其后研究一般认为该反应对大气中HONO的形成不重要[31].最近研究结果表明,光照会保持soot的反应活性,该反应对白天HONO形成可能具有重要作用,尤其是在soot排放量较高的地区[53].

在Ammann等[52]研究基础上,Gutzwiller等[54]发现柴油车尾气排放的半挥发性有机物对HONO形成具有重要影响(R7).柴油车尾气排放的NOχ约2.3%会在半挥发性有机物表面经非均相过程生成HONO. HONO由该途径的生成量至少是柴油车尾气排放量的3倍[54].
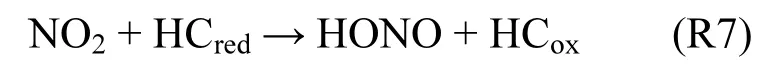
1.3.3 光照催化反应 虽然HONO浓度常在夜间达到最大,但白天HONO也会出现意想不到的高值,这种情况常和光照强度密切相关.许多研究均指出光照会催化某些反应,有利于白天HONO生成.例如,在可见光照射下,NO2在TiO2表面[55]、腐殖酸表面[56]、固态有机化合物表面[57]、酚类物质表面以及地表面[58]都会产生HONO(R8).
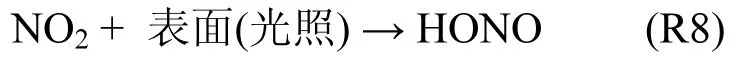
Zhou等[59]发现表面吸附的HNO3或NO3-经紫外光解可以产生HONO,HONO通过解吸作用脱离吸附物表面而释放回大气(R9).Zhou等[60]在森林冠层上空观测到HONO的直接排放,认为沉积到森林冠层表面的 HNO3经光解可生成HONO,该机制有利于已沉积到地表的 HNO3再次活化.Li等[28]指出反应 R9可能是广州后花园白天 HONO的来源之一.Ziemba等[48]在观测实验中发现 HNO3可在一次有机气溶胶表面经非均相过程生成HONO.

2 HONO模拟研究
目前区域空气质量模式,如 CAMχ (http:// www.camx.com/)、CMAQ[61]和 WRF-Chem[62],所采纳的气相化学反应机制主要包括CBM-IV、CBM-Z、SAPRC、RADM2和RACM等[63].以上这些化学机制都将气相反应作为HONO的唯一来源,例如CBM-IV机制中HONO来源包括OH⋅ + NO + M → HONO + M和NO + NO2+ H2O → 2HONO.前文1.2节已指出,NO + NO2+ H2O → 2HONO对HONO的贡献可忽略.由于模式通常不包括HONO的源排放和非均相反应来源,所以HONO模拟值远低于观测值[33,38,50,64].为改进HONO的模拟,Sarwar等[33]将燃烧过程排放HONO、非均相反应R5和光照催化反应R8引入CMAQ模式,HONO模拟结果明显改进,但模拟值仍低于观测值,尤其是在白天时段.Sarwar等
[33]指出非均相反应R5和光照催化反应R8是HONO最主要的两个来源,对HONO的平均贡献分别达54%和32%,而气相反应R2和燃烧过程排放两者对 HONO的贡献仅 14%.非均相反应R5对夜间HONO的贡献高达90%,光照催化反应R8的贡献集中在白天.根据Sarwar等[33]的研究,CMAQv4.7加入了燃烧过程排放HONO、NO2在气溶胶表面和地表面的非均相过程,但尚未详细验证HONO新增来源的影响[65].
Aumont等[66]利用两层箱模式,加入 NO2在soot表面的氧化还原反应 R6.模拟结果表明,若不考虑soot表面的老化过程,HONO模拟值高于观测值.Aumont等[32]进一步考虑了燃烧过程排放HONO以及NO2在颗粒物表面和地表面的非均相反应(R5),发现城市地区冬季夜间HONO浓度比夏季夜间高25%;冬夏两季HONO平均日变化不同,夏季只出现一个峰值,而冬季有两个峰值;污染较重的乡村地区,冬季夜间HONO浓度比夏季夜间高4倍;燃烧过程排放HONO以及NO2在地表面的非均相反应对HONO贡献较大,气溶胶表面的非均相反应贡献较小.
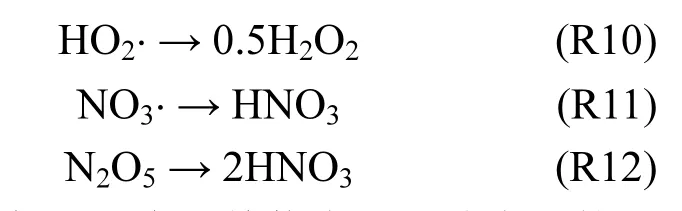
Xu等[67]将气溶胶表面的 4个非均相反应(R5、R10~R12)引入 CMAQ模式,模拟了 2000年6月26~27日北京HONO浓度,模拟值在观测范围之内.Xu等[67]没有考虑气溶胶吸湿性增长过程,北京夏季相对湿度较大,气溶胶吸湿性增长对非均相过程的影响不可忽视(Li等[50]).
Li等[64]将NO2在颗粒物表面和地表面的非均相反应(R5)、NO2在 soot表面的非均相反应(R6)以及 NO2在半挥发性有机物表面的非均相反应(R7)引入WRF-Chem模式,模拟了2006年3月24~29日墨西哥城的空气质量.加入HONO新增来源后,模式可以合理反映HONO的实际日变化;NO2在半挥发性有机物表面的非均相反应(R7)是HONO最重要的来源,对白天HONO的贡献达 75%.因为反应 R7只适用于柴油车尾气排放[54],而Li等[64]将其应用于所有NOχ排放源,可能高估了反应R7的重要性[38].
Volkamer等[68]利用一个箱模式开展了敏感性试验,结果表明光照催化产生HONO的反应,例如NO2在腐殖酸表面[56]的非均相反应,对墨西哥城大气中的HONO并不重要;模式对HONO的汇考虑不充分,致使9:00的HONO模拟值高于观测值70%.Sörgel等[25]模拟结果说明光照条件下NO2在soot表面的非均相反应(R6)[53]对 HONO的贡献<1%,NO2*机制(R4)对HONO的贡献<10%. Li等[50]将HONO源排放(包括燃烧过程排放和反应R7)、NO2*机制以及气溶胶表面的非均相反应进行参数化,并耦合于WRF-Chem模式,HONO模拟结果显著改善.Li等[50]还发现气溶胶表面的非均相化学反应是HONO最重要的来源,贡献约59%;NO2*机制是HONO第二个主要来源,贡献约26%. Czader等[69]指出反应R6与R4对HONO的贡献较小,而NO2的水解反应(R5)可使HONO浓度增大10倍. Gonςalves等[34]在CMAQ模式中加入燃烧过程排放HONO以及NO2在颗粒物表面和地表面的非均相反应(R5),虽然HONO的模拟有很大改进,但仍低于观测值,指出 NO2在地表面的非均相反应参数化方案有待改进.根据世界各地大型观测实验结果,在全球尺度上 Elshorbany 等[11]估算HONO/NOχ均值为0.02,并将该比值应用于一个全球化学输送模式,模拟结果在全球范围内较好反映了 HONO的观测浓度,在高 NOχ排放地区HONO来源对HOχ(≡OH⋅ + HO2⋅)、O3以及PAN(过氧乙酰硝酸酯)有显著影响,并且冬季影响更为显著,建议大气化学模式应考虑HONO新来源,改进HONO化学机制.
3 HONO来源对空气质量的影响
HONO来源影响空气质量的根本原因有两方面.其一,HONO光解产生OH⋅,OH⋅的增加会使大气氧化能力增强,引起O3等二次污染物浓度升高;其二,HONO来源包含多种形成机理,HONO各来源对O3等污染物的影响不同.
Clapp等[70]指出燃烧过程排放HONO对Oχ(≡ NO2+ O3)浓度影响较大.若HONO占NOχ排放量的比例从0%增至5%,O3与Ox的浓度则分别增加 1.51×10-9V/V和 1.15×10-9V/V[71].HONO增加1%可使O3增升约0.3×10-9V/V,是NO2增加1%引起O3增加量的3倍[71].An等[38]认为京津冀地区燃烧过程排放 HONO引起该地区近地面HONO白天和夜间月均浓度最大增幅均超过100%,OH⋅、HO2⋅和O3白天月均浓度最大增幅依次为8%、7%和1%.
关于HONO均相反应来源对O3及相关污染物的影响,近几年关注较多的是 NO2*机制(R4). Wennberg等[72]利用空气质量模式模拟了1987年8月27~29日美国加利福尼亚南部地区O3浓度,结果表明 NO2*机制可使 O3增加 55×10-9V/V, PM2.5增加 20μg/m3.O3模拟值高于观测值,所以Wennberg等[72]认为Li等[42]建议的反应速率常数可能偏大.Sarwar等[73]开展了类似的工作,指出目前美国NOχ排放量远低于1987年,NO2*机制对目前美国空气质量影响很小,但在NOχ和可挥发性有机化合物(VOC)等高排放地区可能影响很大.Ensberg等[74]分别利用美国1987年和2005年源排放清单模拟了 NO2*机制对加利福尼亚南部地区 O3影响,发现单独削减 NOχ排放量时, NO2*机制会促进臭氧浓度下降,这对 O3调控措施有启示意义.Li等[50]指出 NO2*机制可使京津冀主要城市 O3时均峰值最大增量达(30~50)× 10-9V/V,该地区HONO白天月均浓度最大增幅达70%,OH⋅、HO2⋅和O3月均浓度最大增幅依次为19%、16%和4%[38],建议NOχ和颗粒物源排放量偏高地区的空气质量模拟研究应考虑燃烧过程排放 HONO、NO2*机制以及气溶胶表面的非均相化学反应.Jorba等[75]将 NO2*机制耦合到一个全球化学输送模式,模拟结果表明在清洁地区O3浓度可增加(4~6)×10-9V/V,城市地区O3浓度可增加(6~15)×10-9V/V,O3时均浓度最大增量在我国东部可达 30×10-9V/V;NO2*机制在海洋上影响较大,尤其是在 NOχ/VOC比值较大的区域;反应速率常数采用Crowley等[43]的建议值和Li等[42]的建议值对全球大部分地区的模拟结果影响不大,仅对NOχ排放量较高的美国加州、美国东北部、朝鲜半岛以及中国有影响.
关于HONO非均相反应来源对O3及相关污染物的影响,Kotamarthi等[76]将NO2在soot表面的非均相反应R6引入箱模式,发现若不考虑soot表面丧失反应活性,反应R6对大气边界层中O3、OH⋅和 HO2⋅影响显著,O3浓度可增加(8~20)× 10-9V/V;当大气中气溶胶比表面积较大时,反应R6可减少O3夜间损失,使O3夜间浓度升高.由于该箱模式未考虑干沉降和长距离输送等物理过程,Kotamarthi等[76]的结果有待化学和物理过程相互耦合的三维空气质量模式进一步检验以及外场实验的验证.Lei等[77]利用一个化学输送模式讨论了反应R6对O3的影响.在忽略soot还原表面失去活性条件下,反应R6可使O3全天增幅约(4~12)×10-9V/V;O3白天均值增加 7× 10-9V/V,清晨 O3的快速累积过程提早 1小时. Aumont等
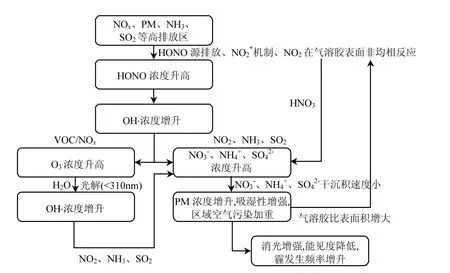
图1 HONO来源对区域能见度及霾影响的概念模型Fig.1 A conceptual model for the impacts of HONO sources on regional visibility and haze
[32]指出HONO来源对O3、NOχ和HOχ的影响在冬季较显著,在夏季影响一般不大,但夏季光化学污染过程除外.Li等[64]将 NO2的非均相反应(R5~7)引入WRF-Chem模式,结果表明O3白天均值增加 6×10-9V/V,清晨 O3的快速累积过程提早2小时,上午二次有机气溶胶的浓度增加了一倍,硝酸盐和铵盐浓度也有所增加,但对硫酸盐浓度影响不大.Xu等[67]在CMAQ模式中加入气溶胶表面的4个非均相反应(R5、R10~12),结果表明反应R5加快了NO向NO2的转化过程,导致O3增加,O3峰值最大增量达67×10-9V/V;反应R5可能是北京城区白天 O3增加的主要原因之一;北京夏季O3主要受VOC控制,反应R5使HONO增多,大气中 OH⋅浓度增加,进而促使 O3浓度升高.Czader等[69]发现反应R5对O3的贡献主要发生在早晨,NO2*机制(R4)和NO2在soot表面的非均相反应(R6)可使白天 O3浓度都有所增加. Gonςalves等[34]指出燃烧过程排放HONO和反应R5可使PM2.5浓度最大增量达14%,主要原因是HONO来源可使硝酸盐浓度增加,并建议空气质量模式加入燃烧过程排放HONO和NO2的非均相反应(R5).An等[38]指出气溶胶表面的非均相反应使京津冀地区近地面HONO白天和夜间月均浓度最大增幅均超过2倍,OH⋅、HO2⋅和O3白天月均浓度最大增幅依次为56%、60%和8%,NO3-、NH4+白天和夜间月均浓度最大增幅均为48%和35%.
4 结论
4.1 HONO来源大体可分为HONO源排放(燃烧过程排放和土壤排放)、均相反应生成(OH⋅与NO的气相反应、机制)和非均相反应生成(水解反应、还原反应和光照催化反应).
4.3 水解反应可能是 HONO最主要来源,空气质量模式模拟结果也支持该观点.不过,是否所有土壤和植被、任何建筑物等表面均可发生该反应仍需大量实验室和外场实验验证.另外,气溶胶表面的非均相反应(水解反应)可能与气溶胶的化学组分有关,不同气溶胶组分的表面反应机率(摄取系数)可能不同,亟需外场实验确定.
4.4 还原反应若光照会保持 soot的反应活性,则在soot高排放地区对HONO贡献较大.光照催化反应仍需大量外场实验证实和定量化.
[1] Levy H II. Normal atmosphere: Large radical and formaldehyde concentrations predicted [J]. Science, 1971,173:141-143.
[2] Finlayson-Pitts B J, Pitts J N. Tropospheric air pollution: Ozone, airborne toxics, polycyclic aromatic hydrocarbons, and particles [J]. Science, 1997,276:1045-1051.
[3] 唐孝炎,张远航,邵 敏.大气环境化学 [M]. 北京:高等教育出版社, 2006.
[4] Platt U, Perner D, Harris G, et al. Observations of nitrous acid in an urban atmosphere by differential optical absorption [J]. Nature, 1980,285:312-314.
[5] Atkinson R, Aschmann S M, Arey J, et al. Formation of OH· radicals in the gas phase reactions of O3with a series of terpenes [J]. Journal of Geophysical Research, 1992,97:6065-6073.
[6] Alicke B, Platt U, Stutz J. Impact of nitrous acid photolysis on the total hydroxyl radical budget during the limitation of oxidant production/Pianura Padana Produzione di Ozono study in Milan [J]. Journal of Geophysical Research, 2002,107,8196,LOP 9-1-LOP 9-17.
[7] Alicke B, Geyer A, Hofzumahaus A, et al. OH· formation by HONO photolysis during the BERLIOZ experiment [J]. Journal of Geophysical Research, 2003,108,8247,PHO 3-1-PHO 3-17.
[8] Stutz J, Alicke B, Neftel A. Nitrous acid formation in the urban atmosphere: Gradient measurements of NO2and HONO over grass in Milan, Italy [J]. Journal of Geophysical Research, 2002, 107,LOP 5-1-LOP 5-15.
[9] Acker K, Febo A, Trick S, et al. Nitrous acid in the urban area of Rome [J]. Atmospheric Environment, 2006,40:3123-3133.
[10] Elshorbany Y F, Kurtenbach R, Wiesen P, et al. Oxidation capacity of the city air of Santiago, Chile [J]. Atmospheric Chemistry and Physics, 2009,9:2257-2273.
[11] Elshorbany Y F, Steil B, Brühl C, et al. Impact of HONO on global atmospheric chemistry calculated with an empirical parameterization in the EMAC model [J]. Atmospheric Chemistry and Physics, 2012,12(20):9977-10000.
[12] An J, Zhang W, Qu Y. Impacts of a strong cold front on concentrations of HONO, HCHO, O3, and NO2in the heavy traffic urban area of Beijing [J]. Atmospheric Environment, 2009, 43:3454-3459.
[13] Spataro F, Ianniello A, Esposito G, et al. Occurrence of atmospheric nitrous acid in the urban area of Beijing (China) [J]. Science of the Total Environment, 2013,447:210-224.
[14] Perner D, Platt U. Detection of nitrous acid in the atmosphere by differential optical absorption [J]. Geophysical Research Letters, 1979,6:917-920.
[15] Matsumoto M, Okita T. Long-term measurements of atmospheric gaseous and aerosol species using an annular denuder system in Nara, Japan [J]. Atmos. Environ., 1998,32(8):1419- 1425.
[16] Hu M, Zhou F M, Shao K S, et al. Diurnal variations of aerosol chemical components and related gaseous pollutants in Beijing and Guangzhou [J]. Journal of Environmental Science and Health, 2002,A37(4):479-488.
[17] Ren X, Brune W H, Mao J, et al. Behavior of OH•and HO2•in the winter atmosphere in New York City [J]. Atmos. Environ., 2006, 40:252-263.
[18] Hao N, Zhou B, Chen D, et al. Observations of nitrous acid and its relative humidity dependence in Shanghai [J]. Journal of Environmental Sciences, 2006,18:910-915.
[19] Lin Y-C, Cheng M-T, Ting W-Y, et al. Characteristics of gaseous HNO2, HNO3, NH3and particulate ammonium nitrate in an urban city of Central Taiwan [J]. Atmospheric Environment, 2006,40:4725-4733.
[20] Qin M, Xie P, Su H, et al. An observational study of the HONO-NO2coupling at an urban site in Guangzhou City, SouthChina [J]. Atmos. Environ., 2009,43:5731-5742.
[21] Kanaya Y, Cao R, Akimoto H, et al. Urban photochemistry in central Tokyo: 1. Observed and modeled OH· and HO2· radical concentrations during the winter and summer of 2004 [J]. Journal of Geophysical Research, 2007,112,D21312,1-20.
[22] Su H, Cheng Y, Shao M, et al. Nitrous acid (HONO) and its daytime sources at a rural site during the 2004PRIDE-PRD experiment in China [J]. Journal of Geophysical Research, 2008, 113,D14312,1-9.
[23] Yu Y, Galle B, Panday A, et al. Observations of high rates of NO2-HONO conversion in the nocturnal atmospheric boundary layer in Kathmandu, Nepal [J]. Atmospheric Chemistry and Physics, 2009,9:6401-6415.
[24] 朱燕舞,刘文清,谢品华,等.北京夏季大气 HONO的监测研究[J]. 环境科学, 2009,30(6):1567-1573.
[25] Sörgel M, Regelin E, Bozem H, et al. Quantification of the unknown HONO daytime source and its relation to NO2[J]. Atmospheric Chemistry and Physics, 2011,11:10433-10447.
[26] Wong K W, Oh H-J, Lefer B L, et al. Vertical profiles of nitrous acid in the nocturnal urban atmosphere of Houston, TX [J]. Atmospheric Chemistry and Physics, 2011,11:3595-3609.
[27] Villena G, Wiesen P, Cantrell C A, et al. Nitrous acid (HONO) during polar spring in Barrow, Alaska: A net source of OH· radicals? [J]. Journal of Geophysical Research, 2011,116,D00R07,1-12.
[28] Li X, Brauers T, Häseler R, et al. Exploring the atmospheric chemistry of nitrous acid (HONO) at a rural site in Southern China [J]. Atmospheric Chemistry and Physics, 2012,12:1497-1513.
[29] Kessler C, Platt U. Nitrous acid in polluted air masses-sources and formation pathways, physico-chemical behaviour of atmospheric pollutants [M]. Reidel, Dordrecht, 1984,412-422.
[30] Kirchstetter T, Harley R, Littlejohn D. Measurement of nitrous acid in motor vehicle exhaust [J]. Environmental Science and Technology, 1996,30:2843-2849.
[31] Kurtenbach R, Becker K H, Gomes J A G, et al. Investigations of emissions and heterogeneous formation of HONO in a road traffic tunnel [J]. Atmos. Environ., 2001,35:3385-3394.
[32] Aumont B, Chervier F, Laval S. Contribution of HONO sources to the NOx/HOx/O3chemistry in the polluted boundary layer [J]. Atmospheric Environment, 2003,37:487-498.
[33] Sarwar G, Roselle S J, Mathur R, et al. A comparison of CMAQ HONO predictions with observations from the Northeast Oxidant and Particle Study [J]. Atmos. Environ., 2008,42:5760-5770.
[34] Gonçalves M, Dabdub D, Chang W L, et al. Impact of HONO sources on the performance of mesoscale air quality models [J]. Atmos. Environ., 2012,54:168-176.
[35] Su H, Cheng Y, Oswald R, et al. Soil nitrite as a source of atmospheric HONO and OH· radicals [J]. Science, 2011,333: 1616-1618.
[36] Kulmala M, Petäjä T. Soil Nitrites Influence Atmospheric Chemistry [J]. Science, 2011,333:1586-1587.
[37] Cleemput O V, Samater A H. Nitrite in soils: accumulation and role in the formation of gaseous N compounds [J]. Fertilizer Research, 1996,45:81-89.
[38] An J, Li Y, Chen Y, et al. Enhancements of major aerosol components due to additional HONO sources in the North China Plain (NCP) and implications for visibility and haze [J]. Advances in Atmospheric Sciences, 2013,30:57-66.
[39] Lu K D, Rohrer F, Holland F, et al. Observation and modeling of OH•and HO2concentrations in the Pearl River Delta 2006: a missing OH· source in a VOC rich atmosphere [J]. Atmospheric Chemistry and Physics Discussion, 2011,11:11311-11378.
[40] Bejan I, Abd-el-Aal Y, Barnes I, et al. The photolysis of ortho-nitrophenols: A new gas phase source of HONO [J]. Physical Chemistry Chemical Physics, 2006,8:2028-2035.
[41] Zhang B, Tao F M. Direct homogeneous nucleation of NO2, H2O, and NH3for the production of ammonium nitrate particles and HONO gas [J]. Chemical and Physical Letters, 2010,489:143-147.
[42] Li S, Matthews J, Sinha A. Atmospheric hydroxyl radical production from electronically excited NO2and H2O [J]. Science, 2008,319:1657-1660.
[43] Crowley J N, Carl S A. OH· formation in the photoexcitation of NO2beyond the dissociation threshold in the presence of water vapor [J]. Journal of Physical Chemistry, 1997,101A:4178-4184. [44] Finlayson-Pitts B J, Wingen L M, Sumner A L, et al. The heterogeneous hydrolysis of NO2in laboratory systems and in outdoor and indoor atmospheres: integrated mechanism [J]. Physical Chemistry Chemical Physics, 2003,5:223-242.
[45] 丁 杰,朱 彤.大气中细颗粒物表面多相化学反应的研究 [J].科学通报, 2003,48(19):2005-2013.
[46] 葛茂发,刘 泽,王炜罡.二次光化学氧化剂与气溶胶间的非均相过程 [J]. 地球科学进展, 2009,24(4):351-362.
[47] Wojtal P, Halla J D, McLaren R. Pseudo steady states of HONO measured in the nocturnal marine boundary layer: A conceptual model for HONO formation on aqueous surfaces [J]. Atmospheric Chemistry and Physics, 2011,11:3243-3261.
[48] Ziemba L D, Dibb J E, Griffin R J, et al. Heterogeneous conversion of nitric acid to nitrous acid on the surface of primary organic aerosol in an urban atmosphere [J]. Atmos. Environ., 2010,44(33):4081-4089.
[49] Notholt J, Hjorth J, Raes F. Formation of HNO2on aerosol surfaces during foggy periods in the presence of NO and NO2[J]. Atmos. Environ., 1992,26(2):211-217.
[50] Li Y, An J, Min M, et al. Impacts of HONO sources on the air quality in Beijing, Tianjin and Hebei Province of China [J].Atmos. Environ., 2011,45:4735-4744.
[51] Zhu T, Shang J, Zhao D F. The roles of heterogeneous chemical processes in the formation of an air pollution complex and gray haze [J]. Science in China, 2011,54B:145-153.
[52] Ammann M, Kalberer M, Jost D T, et al. Heterogeneous production of nitrous acid on soot in polluted air masses [J]. Nature, 1998,395:157-160.
[53] Monge M E, D’Anna B, Mazri L, et al. Light changes the atmospheric reactivity of soot [J]. Proceedings of the National Academy of Sciences, 2010,107:6605-6609.
[54] Gutzwiller L, Arens F, Baltensperger U, et al. Significance of semivolatile diesel exhaust organics for secondary HONO formation [J]. Environ. Sci. Technol., 2002,36: 677-682.
[55] Ndour M, D’Anna B, George C, et al. Photoenhanced uptake of NO2on mineral dust: Laboratory experiments and model simulations [J]. Geophysical Research Letters, 2008,35,L05812, doi:10.1029/2007GL032006.
[56] Stemmler K, Ammann M, Donders C, et al. Photosensitizied reduction of nitrogen dioxide on humic acid as a source of nitrous acid [J]. Nature, 2006,440:195-198.
[57] George C, Sterkowski R S, Kleffmann J, et al. Photoenhanced uptake of gaseous NO2on solid organic compounds: A photochemical source of HONO? [J]. Faraday Discussion, 2005, 130:195-210.
[58] Wong K W, Tsai C, Lefer B, et al. Daytime HONO vertical gradients during SHARP 2009 in Houston, TX [J]. Atmospheric Chemistry and Physics, 2012,12:635-652.
[59] Zhou X, He Y, Huang G, et al. Photochemical production of nitrous acid on glass sample manifold surface [J]. Geophysical Research Letters, 2002,29,1681:261-264.
[60] Zhou X, Zhang N, TerAvest M, et al. Nitric acid photolysis on forest canopy surface as a source for tropospheric nitrous acid [J]. Nature Geoscience, 2011,4:440-443.
[61] Byun D, Schere K L. Review of the governing equations, computational algorithms, and other components of the models-3: Community Multiscale Air Quality (CMAQ) modeling system [J]. Applied Mechanics Reviews, 2006,59:51-77.
[62] Grell G A, Peckham S E, Schmitz R, et al. Fully coupled “online”chemistry within the WRF model [J]. Atmos. Environ., 2005,39: 6957-6975.
[63] 石玉珍,徐永福,贾 龙.大气化学机理的发展及应用 [J]. 气候与环境研究, 2012,17(1):112-124.
[64] Li G, Lei W, Zavala M, et al. Impacts of HONO sources on the photochemistry in Mexico City during the MCMA-2006/ MILAGO Campaign [J]. Atmospheric Chemistry and Physics, 2010,10:6551-6567.
[65] Foley K M, Roselle S J, Appel K W, et al. Incremental testing of the Community Multiscale Air Quality (CMAQ) modeling system version 4.7 [J]. Geoscientific Model Development, 2010,3:205-226.
[66] Aumont B, Madronich S, Ammann M, et al. On the NO2+ soot reaction in the atmosphere [J]. Journal of Geophysical Research, 1999,104:1729-1736.
[67] Xu J, Zhang Y, Wang W. Numerical study on the impacts of heterogeneous reactions on ozone formation in the Beijing urban area [J]. Advances in Atmospheric Sciences, 2006,23:605-614.
[68] Volkamer R, Sheehy P, Molina L, et al. Oxidative capacity of the Mexico City atmosphere-Part 1: A radical source perspective [J]. Atmospheric Chemistry and Physics, 2010,10:6969-6991.
[69] Czader B H, Rappenglück B, Percell P, et al. Modeling nitrous acid and its impact on ozone and hydroxyl radical during the Texas Air Quality Study 2006 [J]. Atmospheric Chemistry and Physics, 2012,12:6939-6951.
[70] Clapp L J, Jenkin M E. Analysis of the relationship between ambient levels of O3, NO2and NO as a function of NOxin the UK [J]. Atmos. Environ., 2001,35:6391-6405.
[71] Jenkin M E, Utembe S R, Derwent R G. Modelling the impact of elevated primary NO2and HONO emissions on regional scale oxidant formation in the UK [J]. Atmos. Environ., 2008,42:323-336.
[72] Wennberg P O, Dabdub D. Rethinking ozone production [J]. Science, 2008,319:1624-1625.
[73] Sarwar G, Pinder R W, Appel K W, et al. Examination of the impact of photoexcited NO2chemistry on regional air quality [J]. Atmos. Environ., 2009,43:6383-6387.
[74] Ensberg J J, Carreras-Sospedra M, Dabdub D. Impacts of electronically photo-excited NO2on air pollution in the South Coast Air Basin of California [J]. Atmospheric Chemistry and Physics, 2010,10:1171-1181.
[75] Jorba O, Dabdub D, Blaszczak-Boxe C, et al. Potential significance of photoexcited NO2on global air quality with the NMMB/BSC chemical transport model [J]. Journal of Geophysical Research, 2012,117,doi:10.1029/2012JD017730.
[76] Kotamarthi V R, Gaffney J S, Marley N A, et al. Heterogeneous NOxchemistry in the polluted PBL [J]. Atmos. Environ., 2001, 35:4489-4498.
[77] Lei W, Zhang R, Tie X, et al. Chemical characterization of ozone formation in the Houston-Galveston area: A chemical transport model study [J]. Journal of Geophysical Research, 2004,109, D12301,doi:10.1029/2003JD004219.
Advances in HONO sources, HONO simulations, and the impacts of the HONO sources on regional or global airquality.
AN Jun-ling*, LI Ying, TANG Yu-jia, CHEN Yong, QU Yu (State Key Laboratory of Atmospheric Boundary-layer Physics and Atmospheric Chemistry, Institute of Atmospheric Physics, Chinese Academy of Sciences, Beijing 100029, China). China Environmental Science, 2014,34(2):273~281
HONO sources (i.e., HONO emissions, homogeneous gas-phase production and heterogeneous reaction production), HONO simulations, and the impacts of the HONO sources on regional or global air quality were reviewed. Reaction of photoexcited NO2with water vapor contributed much to HONO formation in elevated NOχemission areas but the accurate reaction rate needs to be quantified. Heterogeneous formation of HONO on wet surfaces could be the key source of HONO, which was supported by air quality model simulations. The photosensitized NO2reduction on soot is possibly a large contributor to HONO concentrations in high soot emission areas but the related field studies are required. Observations of HONO emissions from soil are very limited and urgently needed.
HONO;hydroxyl radical;NOχ;aerosol;heterogeneous reaction
X131.1
:A
:1000-6923(2014)02-0273-09
安俊岭(1967-),男,宁夏海原人,研究员,博士,主要从事大气环境/大气化学研究.发表论文50余篇.
2013-05-30
国家自然科学基金(41175105);中国科学院战略性先导科技专项(B类)课题“大气灰霾跨界输送途径与定量评估”(XDB05030301)
∗ 责任作者, 研究员, anjl@mail.iap.ac.cn
