动物源性医疗器械中残留DNA表征和风险的研究进展
2023-12-08孙晓霞刘成虎朱艺馨屈秋锦阮文婷
孙晓霞, 刘成虎, 朱艺馨, 屈秋锦, 阮文婷
(山东省医疗器械和药品包装检验研究院, 国家药品监督管理局生物材料安全性评价重点实验室, 济南 250101)
随着生物医药、工程材料等新技术的飞速发展,动物源性医疗器械被越来越多地用于疾病治疗,以实现修复、替代、维持或改善组织和器官功能的目的。这是因为,相较于金属、合成高分子等人工合成材料,动物源性医疗器械不仅具有良好的生物相容性,其广泛的组织来源还能提供类似于人体的三维网状支架结构以利于组织再生修复[1-2]。为了降低针对动物源性成分的免疫风险,在医疗器械的生产过程中常采用脱细胞等工艺,去除细胞等相关免疫原性成分[3-4],但由于DNA具有稳定性,其终产品上仍有可能残留宿主细胞DNA[5-6],因此早在1987年世界卫生组织(World Health Organization,WHO)就将残留DNA含量作为动物源性医疗器械质量控制的一项关键性能指标[7]。
许多研究显示动物源性材料中残留的DNA具有生物活性,尤其当动物源性材料中含有病毒感染细胞或致癌/致瘤潜能的细胞时,其DNA亦可能携带HIV病毒或Ras癌基因等片段[8-10],从而导致病毒感染、致瘤和异常基因表达等安全性风险。同时,风险评估研究认为,DNA片段大小的减小极有可能进一步降低DNA的风险并增加安全裕度,因为残留的DNA片段越小,存在完整癌基因和其他功能序列的可能性就越低[11-13]。由此可见,采用适宜检测方法表征的残留DNA水平对于保证动物源性医疗器械的安全性和质量可控性至关重要。
由于具有学科综合性强、技术含量和附加值高的特点,使动物源性医疗器械产品的研发成为热点。尽管目前关于动物源性医疗器械中可接受残留DNA水平(即DNA限值)的研究仍较少,但国内外各类监管机构已发布了细胞制品等生物源性材料的指导原则和建议要求[14-16]。随着DNA风险研究的深入,监管机构也在不断更新优化残留DNA限值要求,这将有助于确保动物源性等生物材料被科学管理、安全使用。此外,聚合酶链反应(polymerase chain reaction, PCR)等分子生物学技术的发展,使残留DNA的检测方法更加科学、准确,为动物源性医疗器械产品开展合理的质量控制提供了技术保证[17-19]。通过分析动物源性材料中残留DNA的潜在风险,回顾和比较监管机构已发布的生物制品中残留DNA的限值要求和各国药典中收录的残留DNA测定方法;同时结合工艺、材料控制等充分考虑风险,探讨降低DNA残留量的措施手段。旨在推动动物源性医疗器械生产技术水平提高,有效去除器械中残留DNA,通过合理的质量控制,正确进行风险/收益评估,从而保障动物源性医疗器械的安全使用。
1 残留DNA的风险来源和潜在危害
DNA是遗传物质的载体,存储着生物体的形态和功能信息。通过受体介导的内吞作用,机体代谢产生的凋亡细胞DNA和进入机体的外源性DNA能够被哺乳动物细胞摄取,由细胞内核酸酶降解和甲基化酶甲基化而失活,随着细胞分裂被逐渐清理[20-21]。而未被降解的外源性DNA,特别是一些小片段DNA片段(长度约500 bp)则可能进入细胞核、插入基因组[22-23],由此产生的生物活性作用将导致不可预见的安全性风险。由于不同来源的动物源性材料具有不同的潜在风险,并且残留DNA片段大小各异,使其风险复杂多样。1999年美国食品药品监督管理局(U.S. Food and Drug Administration, FDA)在关于“细胞来源疫苗的监管进展”工作组会上,回顾以往生物材料使用史,指出宿主细胞中残留DNA具有致癌性、感染性、免疫原性以及致突变性等风险[24]。
1.1 致瘤性/致癌性
残留DNA致癌的主要机制是引入了显性致癌基因。如果编码癌基因的残留DNA进入细胞核病并激活相应的癌基因,正常细胞会发生转化并分化为瘤细胞。Zheng等[12]、Sheng等[25]的研究已证实表达癌基因H-ras和c-myc的质粒DNA可以在皮下接种后诱导含有这两种癌蛋白的肿瘤形成,并且1 μg的质粒DNA即可诱导新生NIH Swiss小鼠成瘤[9]。
此外,在禽类和哺乳动物中还观察到由于残留DNA插入导致的肿瘤形成[26-30]。然而,由于不同种属间DNA序列的异质性和基因组复杂性[31-32],使得这种插入后发生DNA整合并且整合位置在原癌基因或抑癌基因区域的概率非常低,其中整合并活化一个癌基因的概率是10-10,而原癌基因激活和抑癌基因失活同时发生的概率是10-19[33-34]。因此,相对于插入引起的致癌性,人们更需要关注残留DNA引入的显性致癌基因。对残留DNA的致癌性研究提示人们在选择动物源性材料时,应尽量选取正常的二倍体细胞,避免采用连续传代或具有致癌潜能的细胞,同时也要控制DNA残留量和DNA片段大小,从而尽可能地降低残留DNA的致癌性风险。
1.2 感染性
残留DNA的感染性风险来自于细胞中存在的感染性病毒基因组。研究发现许多病毒基因组具有感染性,例如人乳头瘤病毒(HPV)[35]、腺病毒[36]、疱疹病毒[37]、EB病毒(EBV)[38]、乙肝病毒(HBV)[39]等,通过将病毒DNA整合到宿主细胞基因组上而产生感染性病毒。除了DNA病毒,外源性逆转录病毒也具有感染性风险,如来自鸟类和啮齿动物的逆转录病毒(迄今为止还未发现人类内源性的逆转录病毒具有感染性)。由于宿主细胞中存在的多种感染性病毒基因组,可见DNA感染性风险可能比致癌性风险更高。Sheng-Fowler等[8]、Chabot等[40]对HIV病毒DNA的研究表明,线状DNA的感染力是环状DNA的30~100倍,1 pg残留线状DNA就具有感染性,而HIV感染细胞中环状DNA的致病剂量为2 μg。考虑到残留DNA的病毒感染性风险,除了对动物源性材料中病毒进行灭活,人们还需要从残留DNA的数量和片段方面来降低病毒感染性。
1.3 免疫原性
尽管DNA分子缺乏固定的空间结构、不能作为抗原被抗体识别,然而当DNA被设计成编码某种蛋白质抗原的重组真核表达载体,即作为DNA疫苗,外源基因可以在体内表达,产生的抗原能够激活机体免疫系统而用于免疫治疗[41]。但许多研究报道,DNA免疫时可在注射局部引起非治疗效应的炎症反应[41-42]。这可能与DNA诱导局部免疫应答而介导的免疫刺激作用有关,例如根据Hornung等[43]的研究结果,细胞中的DNA能够与酶结合形成复合物而激活天然免疫系统,释放炎症小体引起发热和局部肿胀等炎症症状。此外,Mostafa等[44]研究发现自闭症儿童的血液中抗双链DNA(ds-DNA)抗体阳性率(34%)明显高于健康儿童(2%),提示这些抗体可能参与自闭症儿童的自身免疫攻击。若动物源性材料中残留的DNA与儿童自身的DNA序列相似或重叠,尽管这种情况的发生概率极低,但当儿童暴露于这种非自身DNA片段时,则可能会产生与其自身DNA相交叉的免疫反应,诱发机体免疫损伤。
1.4 致突变性
DNA插入基因组后还可能导致基因突变。研究发现X1-连锁严重联合免疫缺陷病(SCID-X1)患者接受基因治疗后,常发生基因易位和突变等异常而导致治疗失败[45-47],提示外源性DNA片段可能被外周血干细胞摄取、引起基因突变。当突变发生于胶质细胞时,则可能导致神经系统疾病。例如,DNA双链断裂(double strand break, DSB)不仅见于紫外线、电离辐射以及烷化剂等化学性损伤,还是神经活动中一个关键且正常的过程并能被机体快速修复[48]。研究显示,参与DSB修复的基因在自闭症中发生了突变,导致错误的基因重组或修复失败[49-50]。此外近来研究发现,精神分裂症等神经发育障碍性疾病与保护性基因缺失或异常导致的γ-氨基丁酸(γ-aminobutyric acid,GABA)能信号系统功能失调密切相关[51]。这些由DNA插入诱发的基因突变研究提示,尽管外源性DNA插入基因组并在特点位点整合的可能性很低[52-54],但对于本就具有基础性疾病的患者或DNA代谢活跃的人群,由外源性DNA插入导致的基因突变风险将极大增加,引起基因重组异常或修复失败以及多个组织器官功能障碍。
2 关于残留DNA的法规要求
动物源性材料作为一个快速发展的新兴技术领域,目前尚无此类生物材料中残留DNA规定和要求,有关残留DNA的基本法规均来自疫苗等生物制品。在生物制品的生产过程中,由于细胞基质残留的DNA可能存在致癌性、感染性等安全性问题,因此各监管机构(WHO、FDA及NMPA等)对残留DNA进行了严格把控,以保证生物制品的安全使用。这里汇总了过去数十年,国内外监管机构出台的生物制品中可接受的残留DNA水平的法规要求。
2.1 国外
细胞等生物材料中残留DNA的安全性风险主要与其潜在致癌性有关,这是由于DNA插入基因组后可能发生癌基因激活。但最初由于研究数据有限,世界卫生组织(WHO)根据Vero细胞来源的脊髓灰质炎疫苗中DNA含量,采用10 pg/剂作为疫苗中残留DNA限量[55]。之后,随着致癌性相关实验数据的增多,研究人员假设在体内1 ng DNA对应的活化癌基因约为100个拷贝,由致癌性风险评估得出机体发生致癌性转化的概率为10-9[56]。由此WHO的生物制品工作组推断,100 pg DNA约对应1个拷贝的活化癌基因,发生致癌性转化的概率为0.5×10-10且该风险是可以忽略的,于是在1986年WHO将残留DNA的限值提高为100 pg/剂[7, 57]。然而该限值仍较严格,尤其是对于某些病毒疫苗,例如某些包膜病毒常常难以符合限值要求,由此导致相关疫苗的生产成本增加、价格上涨甚至造成严重的市场短缺。进一步的癌症研究发现,DNA诱导致癌还与基因突变、表观遗传等多种机制密切相关。此外,对于连续传代的非致癌细胞,如常见的Vero细胞等,这些细胞中不太可能存在活化的显性癌基因[58-60]。因此WHO在1997年组织专家再次讨论并修改了DNA的限值,并认为减小残留DNA片段大小有可能进一步降低致癌性、感染性等DNA的风险[61]。
目前WHO、美国FDA和欧洲药典建议,生物材料的风险评估时应考虑原料细胞的性质和给药途径。WHO和美国FDA推荐,来源于连续传代非致瘤细胞的生物制品,当胃肠道外使用时,无论是否用于婴幼儿,其残留DNA应不高于10 ng/剂,长度不大于200 bp[24, 62-63]。同时,基于DNA经口和胃肠道暴露的风险差异,FDA建议将口服类疫苗的DNA残留量放宽至100 μg/剂;以人二倍体细胞系来源(如被广泛用于生物制品的MRC-5细胞和WI-38细胞)的生物制品,FDA不认为其残留DNA具有安全性风险[64]。相对于WHO,欧盟提高了婴幼儿使用疫苗的要求,婴幼儿疫苗≤100 pg/剂,非婴幼儿疫苗≤10 ng/剂。欧洲药典10.0根据最小的功能片段长度,认为<200 bp的DNA是安全的[16]。
2.2 中国
中国药典2020年版三部规定,以大肠杆菌或真菌基质生产的疫苗和治疗性生物制品,其DNA残留量不能超过10 ng/剂;对于以细胞基质生产的灭活疫苗和治疗性生物制品,其DNA残留量应低于100 pg/剂[14]。同时,药典2020年版还优化了安全性检测方法,除了DNA探针杂交法和荧光染色法,在通则3407“外源性DNA残留量测定法”中新增了“定量PCR法”,通过进一步完善生物材料的质量控制方法,将人用狂犬病疫苗的DNA残留量由100 pg/剂调整为3 ng/剂,然而以CHO细胞为来源的重组乙型肝炎疫苗仍保留10 pg/剂的限值要求[17]。中国现行2020版药典规定检测DNA残留量的疫苗、生物制品及标准汇总分别如表1(疫苗)和表2(生物制品)所示[14]。
3 残留DNA的测定方法
基于动物源性医疗器械的安全性和质量可控性,建立准确、灵敏且特异性强的残留DNA测定方法是至关重要的。各国药典给出了数种检测方法用于表征残留DNA,包括DNA探针杂交法、荧光染色法定量PCR法以及基于DNA结合蛋白免疫酶法[15-17, 65-66]。这些方法可用于在产品生产过程中和成品上测定DNA残留量和DNA片段大小。
3.1 DNA探针杂交法
供试品中的DNA经变性为单链后吸附于固相膜上,在一定条件下(适宜的温度及离子强度等)与特异性标记的单链DNA探针退火复性、杂交形成双链DNA,使用与标记物相应的检测系统显示杂交结果,颜色深度与DNA含量相对应,通过与已知含量的阳性DNA比对,计算供试品中的DNA残留量[15, 17]。DNA探针杂交技术,将已知的DNA 序列片段作为杂交探针,通过放射性或荧光标记,能够特异性检测未知的DNA样品片段及其相对大小。然而在实际操作时,放射性标记物(如32P)存在半衰期短、放射等问题,而荧光标记的探针如果采用仪器读取信号,杂交法理论上可以达到定量检测要求的灵敏度,但是检测时间需要48 h;由此可见,这种方法的操作比较烦琐、费时,操作过程中残留的溶剂可能会干扰检测结果,这些不足限制了杂交法的应用。
3.2 免疫阈值法
供试品中DNA变性为单链DNA(ssDNA)后,能够同时与链霉亲和素标记的ssDNA结合蛋白和脲酶偶联的抗ssDNA单抗结合,形成含有DNA-链霉亲和素-脲酶的复合物并被生物素化膜捕获。在尿素溶液中,膜上的复合物通过与尿素反应产生氨(NH3),引起溶液pH改变且改变与DNA含量成正比[15-16]。因为是采用DNA抗体的非特异序列免疫检测技术,因此阈值法不能特异性识别宿主残留DNA序列,且容易受到环境和操作人员的DNA污染,导致读值偏高。
3.3 定量PCR法(qPCR法)
针对供试品DNA设计特异性引物和带有荧光标记的探针,在PCR反应过程中,通过荧光标记探针或掺入荧光染料,连续监测反应体系中荧光数值的变化,实时反映特异性扩增产物量的变化。当PCR反应释放的荧光强度达到预设的阈值时,体系的PCR循环数(Ct值)与该体系所含的起始DNA模板量的对数值呈线性关系;采用已知含量的DNA标准品构建标准曲线,通过Ct值计算供试品中的DNA残留量[15-17]。qPCR法具有序列特异性,其灵敏度、准确度、精密度等性能好,可以实现快速、高通量DNA片段检测;其中,特异且高度保守的q-PCR引物/探针等试剂是保证残留DNA准确可靠检测的关键。

表2 中国药典规定的治疗性生物制品中DNA残留量标准[14]Table 2 The limits of residual DNA in therapeutic biologics according to provisions for Chinese Pharmacopoeia[14]
3.4 荧光染色法
这种方法仅适用于测定双链DNA。双链DNA荧光染料(PicoGreen)与供试品中的双链DNA特异结合形成复合物,在波长480 nm激发下产生荧光信号,使用荧光酶标仪在波长520 nm处进行检测;在一定DNA浓度范围内,荧光强度与DNA浓度成正比,可采用已知含量的双链DNA标准品构建标准曲线,根据荧光强度计算供试品中的DNA残留量[17]。除了双链DNA,动物源性医疗器械中还可能残留单链DNA片段,且残留的DNA片段大小不一,而这种方法中仅仅是将已知含量的双链DNA作为标准品,这些极大的影响了残留DNA检测的准确性。
在选择合适的残留DNA检测方法时,需结合样品的特征考虑检测方法的特异性和局限性(见表3)。其中,qPCR方法同时被中国药典[17]、美国药典[15]和欧洲药典[16]收录,其技术优势在于序列特异性高、灵敏度高、重现性好,还可以同时实现定量和相对分子质量的高通量检测,使得操作更简便、结果更精确,从而为企业在工艺研究和成品质量控制方面提供了可靠的检测手段。而现行版中国药典收录的以PicoGreen为代表的荧光染料法[17],由于存在技术缺陷、不能准确定量,已经被美国和欧盟药典摒弃。
4 降低DNA风险的手段
对于动物源性医疗器械中残留DNA的风险,控制其DNA生物活性是关键。除了减少DNA残留量,对于胃肠外使用的生物制品,由于残留的DNA中可能包含致癌基因,WHO还建议将DNA片段化,使其大小降低到功能基因以下,即小于200个碱基对[63]。目前常用的降低DNA残留量及其大小的方法包括的酶解、渗滤、超滤、层析等[67-69]。在生物材料中加入核酸酶,如DNase Ⅰ、非限制性核酸内切酶等,可以靶向DNA将大分子酶解成小片段并降低了其分子黏度,但这种方式产生的DNA片段大小不一,还会导致酶的残留。根据DNA带有负电荷的特点,可使用富含正电荷的物质,如聚乙烯亚胺(polyethyleneimine, PEI)、鱼精蛋白等与之中和形成沉淀而去除DNA[70];也可以采用阴离子交换剂或亲和层析的方式,通过控制吸附、洗脱条件来除去带负电荷的DNA[71]。尽管利用电荷来去除DNA的操作简便,但不适用于同样带有负电荷的材料;若产品带正电荷,还会与DNA发生聚集和包裹,导致DNA去除效果不理想[72]。此外,也可采取化学灭活的方式,如β-丙内酯(BPL)等,降低产品中病毒DNA的大小和活性。
然而动物源性医疗器械生产过程中的一般工艺难以有效去除DNA,这一方面是由于DNA超强的化学稳定性,另一方面是由于DNA呈负电荷易与其他生物大分子结合从而产生聚集(或吸附)、包裹作用。考虑到不同的组织来源和用途,可以通过多种去除方式组合的方案,以实现更加充分的DNA去除效果。
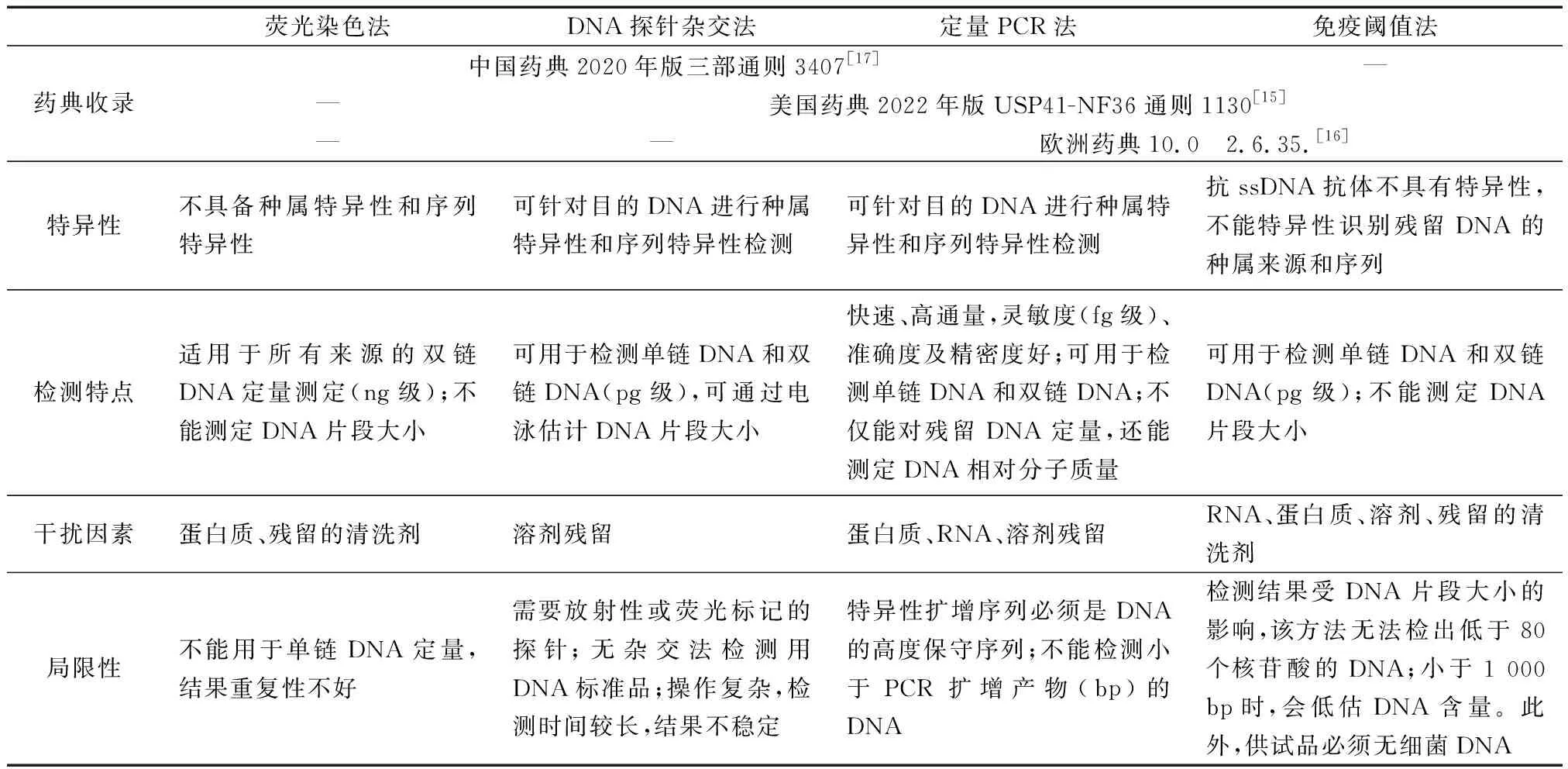
表3 不同DNA残留量检测方法异同Table 3 Comparison of the methods for residual DNA testing
5 总结与展望
随着越来越多的动物源性医疗器械参与疾病治疗,对其质量控制也日益严格。对于去除免疫原性物质的医疗器械,尽管在制造过程中进行了脱细胞等工艺,但最终产品中仍可能残留DNA片段。因此,动物源性医疗器械中残留的DNA同样存在安全性风险和需要开展风险管理。
基于风险分析的安全性研究是进行产品质量控制的基础,通过分析动物细胞残留DNA的潜在风险和来源,追溯了生物制品中残留DNA可接受水平的演变,显示该限值是基于不同材料来源和使用条件下的残留DNA风险评估过程。尽管相较于生物制品,动物源性医疗器械与人体的接触方式更多样,如表面接触、外部接入、植入等[73],但它们都来自于生物材料且不可避免的残留了宿主细胞DNA,这提示针对动物源性医疗器械中残留DNA开展风险管理时,除了分析这些DNA可能编码或隐藏的癌基因或感染因子等潜在危害,还应关注不同类型的医疗器械,通过深入探究不同接触方式、接触周期器械的安全性问题,为科学监管提供残留DNA表征和稳健风险评估等数据支持,从而保障此类器械的安全使用。
为了尽可能去除外源污染、确保产品质量,对宿主细胞残留DNA的表征和测试是控制动物源性医疗器械生产的一个关键组成部分。这些残留DNA的生物活性决定了其安全性风险,它与DNA的多少和片段大小密切相关。FDA等的监管指南建议胃肠外使用生物制品中残留DNA水平为10 ng/剂和200个碱基对大小,而中国药典仅对残留量的限值进行了要求(如表1和表2所示)。由于DNA片段大小与其功能活性密切相关[8-10, 22-23],并且风险评估的研究表明,相同质量DNA的条件下,DNA片段的减小将有助于降低风险[11-12]。这说明除了DNA含量,在动物源性医疗器械产品进行质量控制、提高安全性时,还应关注残留的DNA片段大小。
残留DNA表征的目的包括:①确认纯化工艺合理,能有效去除宿主DNA残余;②确认产品中杂质含量符合标准要求。非特异性的DNA检测结果不能区分究竟是生产中污染、检测污染、还是工艺缺陷引起的DNA残留,所以无法为解决方案提供有效信息。在严格的生产体系中,残留DNA准确表征能够解决工艺合理性问题。为了加强对产品生产的过程监测和质量控制,各国药典都对残留DNA的检测方法进行了更新,通过提高检测方法的敏感性确保产品中残留DNA的安全性。中国药典中的检测方法与美国、欧洲各不相同[15-17]。因此,中国相关器械企业在研发与生产中应具有前瞻性,尽早更新相关检测方法,这将有利于改进工艺、提高产品质量,有利于产品走出国门提高产品的竞争力。
动物源性医疗器械的风险评估是基于风险-收益权重的产品安全性和有效性分析过程。尽管监管机构和生产企业将残留DNA作为进行产品质量控制的性能指标,但任何生物过程中残留DNA的水平还是关键的安全性风险,为制造过程和产品上市提供安全性保证。对于动物源性医疗器械中残留DNA带来的风险隐患,采用安全可靠的工艺手段去除DNA是降低风险、保证产品质量的关键。建立合适的残留DNA检测方法有助于监测工艺质量,确保动物源性医疗器械产品的安全性和有效性,造福人类健康。
