超高效液相色谱法测定聚丙烯中成核剂
2023-09-05张攀攀姜健准曹雪君
张攀攀,刘 静,姜健准,曹雪君
(中国石化北京化工研究院,北京 100013)
0 前言
PP 是市场上占有量较大的通用塑料之一,为了提高PP 的性能,在制备过程中通常添加多种添加剂如抗氧化剂、光稳定剂、抗静电剂、增塑剂、成核剂等改性。添加剂在塑料中的含量在0.01 %~0.5 %(质量分数,下同)之间[1⁃3],在实际应用中加入微量添加剂对PP 性质有较大影响。成核剂可以改变PP 的结晶特性,加快结晶速率,提高结晶温度和减小球晶的尺寸,是目前最常用的提高PP力学性能、透明度的方法[4⁃5]。
本研究选取了市场上最常见的5 种有机成核剂进行研究,通过双螺杆挤出PP 造粒制备标准样品,将标准样品在与实际样品相同或接近的条件下做前处理,对比了加热提取,超声提取和MAE 提取等3 种前处理方法的优劣性并总结出最优的提取条件,优化了UPLC 条件,将优化的方法用于实际生产中PP 材料中成核剂的定性定量。
1 实验部分
1.1 主要原料
甲醇(MeOH)、乙腈(ACN),LC⁃MS 级,北京百灵威科技有限公司;
异丙醇(IPA)、二氯甲烷(DCM),梯度级,北京百灵威科技有限公司;
乙酸乙酯(EAC),分析纯,北京百灵威科技有限公司;
实验用水,超纯水,默克化工技术(上海)有限公司;
成核剂,M3905、HX⁃3、NX8000,分析纯,美国美利肯化工集团公司;
成核剂,NA21,分析纯,呈和科技股份有限公司;
苯甲酸钠(SOD BENZOATE),分析纯,北京百灵威科技有限公司;
PP粉料,MT20,中国石化石家庄炼化分公司;
5种成核剂的性质见表1。

表1 5种成核剂的性质Tab.1 Properties of the five nucleating agents
1.2 主要设备及仪器
双螺杆挤出机,Polylab OS RheoDrive 7,德国Haake Technik Gmbh公司;
研磨机,A11,德国IKA仪器设备有限公司;
超高效液相色谱仪,配备光电二极管阵列(PDA)检测器,沃特世科技(上海)有限公司;
微波辅助提取仪,JUPITER,上海新仪微波化学科技有限公司;
超声波清洗仪,KQ5200E,江苏省昆山市超声仪器有限公司;
电加热炉,SXKW,北京中科奥博科技有限公司。
1.3 样品制备
标准样品制备:将PP 粉料与成核剂预混合均匀,倒入双螺杆挤出机制备PP 颗粒,双螺杆挤出机5 段温度为190、200、210、210、200、180 ℃。各种成核剂的添加量分别选取了低(0.1 %)、中(0.2 %)、高(0.5 %)3 种添加浓度,并将中等添加量的PP 作实验条件预探索,再考察3 种添加量下的最大提取效率;聚丙烯标准样品颗粒采用液氮冷冻3 min,在12 000 r/min 的研磨机里粉碎为270 μm的粉末,进行后续提取步骤;
PP中成核剂提取方法比较了加热回流提取、MAE提取和超声提取的效果,每次称取1 g样品(精确到0.001 g)于25 mL圆底烧瓶,加入20 mL试剂摇匀后进行提取实验,提取后过滤,用提取剂清洗并定容至25 mL,最后经0.22 μm滤膜后上机检测;每组实验平行测定6次。
2 结果与讨论
2.1 色谱条件优化
实验中选择的成核剂为山梨醇类、磷酸盐类、苯甲酸盐类中常用的几种,分子性质相差大,分离比较困难,故色谱条件的优化非常重要,实验发现 ACQUI⁃TY BEH C18(50 mm×2.1 mm,1.7 μm)柱对烯烃助剂有较好的分离效果。柱温箱35 ℃;流动相B 相为超纯水,C相为乙腈;流速0.3 mL/min。流动相选择梯度洗脱程序能显著缩短检测时间:初始流动条件为30 %B 相和70 % C 相,0~11 min 内C 相线性升至99 %,11~15 min 保持不变以继续洗脱色谱柱上其他可能存在的添加剂;每次进样量1 μL,PDA 检测器波长为各种成核剂均有较大紫外吸收的213 nm。在所选条件下6种成核剂的分离效果如图1所示。

图1 成核剂的保留时间Fig.1 Retention time of the nucleating agents
2.2 外标曲线
实验中分别称取5种成核剂标准品各15 mg(精确至0.1 mg)于50 mL 容量瓶中,以异丙醇溶解定容,得到300.0 mg/L的溶液。分别移取适量标准溶液,用异丙醇逐级稀释为100.0、50.0、20.0、10.0、5.0、1.0 mg/L的混合溶液,采用外标法定量,计算成核剂保留色谱峰面积与对应浓度的关系,结果见表2。将信噪比大于3时的质量浓度作为成核剂的检出限(LOD),信噪比大于10时作为定量限(LOQ),经测定,5种成核剂的检出限和定量限分别在0.1~0.5 mg/L 和0.5~1 mg/L,相关系数均大于0.995,说明各标准曲线均有良好的线性关系。

表2 各组分的外标曲线Tab.2 External standard curves of each component
2.3 前处理方法优化
2.3.1 提取溶剂的选择
所检测的成核剂分子极性差别较大,如M3905、HX⁃3、NX8000 等山梨醇类成核剂属中等极性化合物,苯甲酸钠、NA21 属中等偏强极性化合物,在不同极性的有机溶剂中溶解性差别较大,导致提取效率存在差异。本实验考察了常用的提取溶剂以及它们的混合溶剂为有机提取溶剂时的提取效果。MAE 条件:温度80 ℃,时间10 min,功率500 W。超声提取条件:温度70 ℃,时间30 min,功率300 W。加热回流条件:各溶剂沸点加热,时间4 h。
MAE本质是外加高频电磁波(3×102~3×105MHz)穿透待处理样品内部,使其在微波能激发下升温,样品内部目标提取物快速与基质分离,扩散到溶剂中。如图2(a)所示,MAE 时单一溶剂提取在所设条件下提取效率从高到低分别是乙酸乙酯、异丙醇、乙腈、甲醇。且各溶剂对5 种成核剂提取效率均低于40 %,达不到定量分析需求。提取效率低的限制原因有两方面,一是MAE 对极性大的溶剂才有更好的响应,二是所选成核剂在溶剂中的相似相溶。成核剂属于中等极性化合物,不适合用强极性溶剂提取,这与MAE 对溶剂的选择是矛盾的。二氯甲烷与异丙醇极性远小于甲醇或乙腈,更适合中等极性溶剂提取。1/1混合(体积比)使得提取效率较纯异丙醇高4倍以上,这得益于二氯甲烷对成核剂有较好的溶解性和介电常数较大的异丙醇(介电常数17.9)对微波加热也有很高的响应,兼顾了溶剂与溶质的相溶性和与微波原理的匹配性。故确定二氯甲烷/异丙醇1/1 (体积比)为首选提取剂。

图2 提取溶剂对提取效率的影响Fig.2 Effect of extraction solvent on extraction efficiency
超声提取法根据机械波施加到样品上会产生空化作用,在液体中产生的真空气泡在被提取物表面爆裂,产生局部高温高压,这种高温高压对溶质的影响较大,加快了物质迁移出表面的速率。使用超声提取时[图2(b)]发现乙酸乙酯对山梨醇类成核剂在几种提取剂中效率最高,相同条件下比MAE 的最佳溶剂二氯甲烷与异丙醇按体积比1/1混合高15 %以上,这与MAE的结果有较大差异,这是因为2 种提取方法的原理差异较大,乙酸乙酯在所选溶剂中极性最弱,与山梨醇类成核剂相溶性较好。而对于成核剂苯甲酸钠和NA21,极性较大的异丙醇有更高的提取效率。
加热回流与以上2种提取方法比较,影响因素则更为单一,只取决于溶剂对溶质的相似相溶。为了加快提取速度,图2(c)各溶剂均在沸点下加热,由于异丙醇在几种溶剂中沸点最高,故在相同时间下提取效率也是最高的。由图2(c)可见二氯甲烷与异丙醇按体积比1/1 混合提取效率最低,这是因为二氯甲烷非极性较强,对成核剂溶解性差,与异丙醇混合后使提取效率下降。同时与以上2种提取方法比较,还发现在较长提取时间下各种溶剂提取效率均有显著提高。
综上分析,MAE 用二氯甲烷与异丙醇按体积比1/1混合提取成核剂,超声提取时用乙酸乙酯提取山梨醇类成核剂,异丙醇提取苯甲酸盐和磷酸盐类成核剂。加热回流提取选择异丙醇作为提取溶剂。
2.3.2 提取方法对比
选择2.3.1 节所述不同提取方法中对应的最佳溶剂,比较在相同温度和时间下MAE 和超声提取的提取效率。加热回流提取在低温、短时间内提取效率显然非常低,故选取在异丙醇沸点下加热回流6 h 与前2 种方法对照。实验中对各种样品做5 次平行提取,以1 g样品中所含成核剂质量计算测试结果的极差和标准偏差,以验证提取方法的精密度,结果见表3,由表可知,5 种成核剂提取效率的极差(range)为0.07~0.25 mg/g,相对标准偏差(RSD)在0.5 %~6.7 %之间,其中最大极差出现在MAE 法中,因为MAE 缺少搅拌溶剂的方法,在提取过程中会产生不均匀受微波辐射情况,导致多次平行提取间精密度较差。加热回流提取沸腾状的溶剂带动样品混合,且长时间的提取导致样品被处理非常充分,故精密度最好。超声提取法中超声对溶剂的机械振动类似搅拌的效果,也能充分提取其中成核剂,精密度也较好。

表3 提取方法精密度Tab.3 Precision of the extraction methods
将提取效率平均值作为比较对象,结果如图3 所示,10 min 超声提取效率较MAE 低10 %以上,实验中发现超声提取法在提取30 min 后继续增加时间对提取效率的贡献越来越小,达到MAE 同等效率一般需要多3~6倍的提取时间,可见MAE 法在提取时间上优于超声提取。6 h 加热回流提取效率除苯甲酸钠外均能达到90 %以上,在足够的提取时间下传统提取方法效率接近100 %且能对各种成核剂无选择性提取。故在后续实验中以MAE 作为前处理手段,以加热回流结果作为最大回收率参比。

图3 提取方法比较Fig.3 Comparison of the extraction methods
综合比较3种提取方法,以标准样品的提取效率为主要对象,同时也考虑到方法的操作简便性、处理量和精密度等次要特征,发现MAE 具有显著处理时间短的优点,在相同处理温度和时间下效率也在3种方法中最高,实验中较加热回流提取可节省溶剂,在较为自动化的仪器上操作可大批量处理样品,降低时间成本。MAE 较大的不足在于对使用的溶剂有较大限制,对介电常数小于14 的溶剂不能单独使用,必须添加极性更强的溶剂混合,另外MAE 会产生局部高温等非正常现象,导致精密度在所选3种提取方法中最低。
超声提取重现性好,与加热回流精密度均较高,是一种稳定的前处理方法。用时较加热回流提取短,提取温度适中,与加热回流一样均有很高的提取效率和广泛的溶剂选择。加热回流提取最为稳定,在较高温度下提取数小时能得到理想的提取效率,溶剂用量最大。根据以上优缺点对比取舍,在后续实验中以MAE作为前处理手段,以加热回流结果作为最大回收率参比,优化得到最佳的前处理条件。
2.4 MAE最佳提取条件
2.4.1 MAE温度选择
在提取时间10 min 条件下考察了不同温度下的提取效率,提取时间分别为50、60、70、80、90 ℃,5 种成核剂提取效率随温度变化的规律如图4(a)所示,整体上随着温度升高,各成核剂提取效率随之增大,达到80 ℃后出现了3种情况,NA21和NX8000效率基本不变,说明该条件下2 种成核剂提取效率已经达到最大;5 种成核剂中提取效率最低的苯甲酸钠提取效率仍在增大,表明完全提取苯甲酸钠需要更高的温度;M3905 在温度增加到90 ℃后提取效率不增反降,可能是在较高温度的微波条件下M3905发生了部分消解。本实验的微波装置使用的是一种容量为50 mL 的密封聚四氟乙烯罐,故在瓶中增加温度时同时会增加罐中压力,提高提取溶剂的泡点,达到了部分加速溶剂提取特有的高温高压提取效果,故在低温时升温有利于提取效率提高。实验发现当罐内温度高于90 ℃,一段时间后PP样品基质会发生溶胀,导致提取溶剂被吸附,无法将溶液与基质分离开,同时较高的温度会增加仪器负荷,降低运行寿命,故初步选择提取温度80 ℃左右较为合适。

图4 MAE温度及时间对提取效率的影响Fig.4 Effect of MAE temperature and time on extraction efficiency
2.4.2 MAE时间选择
成核剂在PP 中形成结晶中心,球晶从其表面生长,PP 链与成核剂的结合力较其他添加剂更强,迁移速率慢,难从PP 内部迁移到表面,故需要充足的处理时间。将PP 在80 ℃,DCM/IPA 体积比1/1 条件下分别提取3、6、10、15、20 min,PP 中成核剂的提取效率如图4(b)所示,提取时间的影响与温度对提取效率的影响有相同的响应规律,可见除苯甲酸钠外其他4种成核剂提取效率在6 min 后基本保持不变,说明MAE 能对样品进行快速前处理。苯甲酸钠提取效率随时间增加而持续增大,表明对该种成核剂前处理比其他成核剂需要更多的时间和更高的温度,其提取最难的原因可能为苯甲酸钠与PP 链结合力较强,具体原因有待更深入研究。进一步实验表明,在80 ℃下MAE 时间超过30 min,PP 同样会发生溶胀,吸收溶剂难以洗脱,大大增加了实验误差。故优化前预选择MAE 时间10 min即可。
2.4.3 最佳提取温度时间优化
响应面法能准确直观地反映多个变量与一个响应值之间的函数关系。温度、时间、溶剂用量等对提取效率有显著影响,故根据响应面法的中心组合设计(CCD)优化探索最佳条件,能得到比单因素实验更准确全面的结果,实现对一种成核剂确定一个最佳的MAE条件,以满足实际样品定量分析需求。CCD初始值采用Design⁃Expert 软件获取,设定温度范围为60~90 ℃,时间选择3~15 min,溶剂用量为15~25 mL/g,获得的实验数据及结果如下表4 所示,由CCD 估计的成核剂M3905 的响应面如图5 所示,预测结果显示提取温度83 ℃,时间13 min 足以获得最大的提取效率。其他各组标准样品也得到了相似的结论,结果总结于表5。选择优化的条件进行精密度实验。

图5 Design⁃Expert预测M3905提取响应三维图Fig.5 3D map of extraction response predicted by Design⁃Expert for M3905
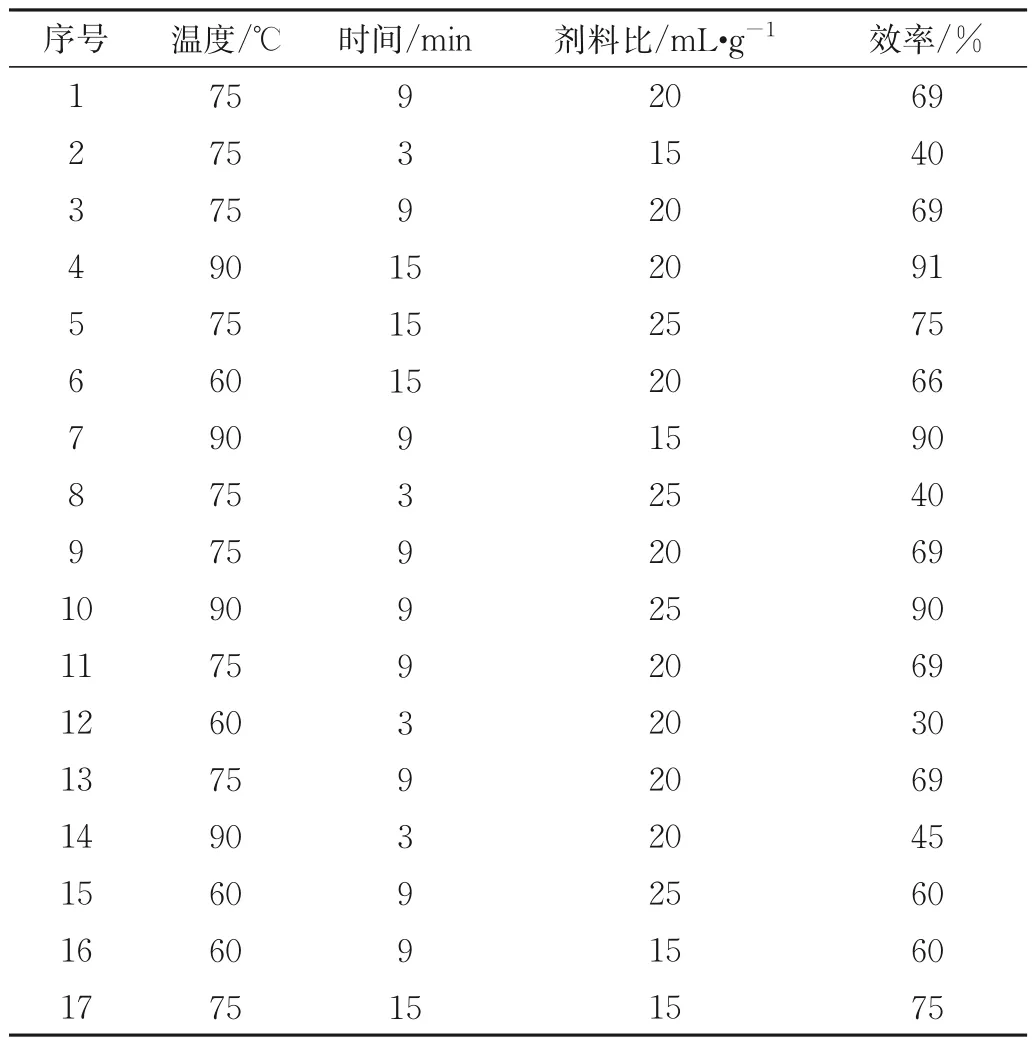
表4 CCD因素水平Tab.4 Factor level of CCD

表5 Design⁃Expert预测的MAE最佳条件Tab.5 Optimal MAE conditions predicted by Design⁃Expert
2.5 回收率及相对标准偏差
本实验参照了市面上PP 中成核剂的常见添加量,设置了低、中、高3 种添加水平,将不同添加量的PP 颗粒进行了回收率和精密度实验,每个样品在Design⁃Ex⁃pert预测的最佳条件下做3次平行MAE 实验后各测定3 次,计算回收率平均值和相对标准偏差,结果见表6,最佳条件下回收率在83.4 %~113.1 %之间,符合国家标准GB/T 27404—2008 中对微量物质加标回收率80 %~120 %的允许限,其中HX⁃3 加标回收率高于100 %,可能为偶然误差和人为误差导致。本次RSD(n=9)在0.9~7.9之间,方法精密度可以满足PP 中微量成核剂的分析需求。

表6 5种成核剂的回收率和相对标准偏差(n=9)Tab.6 Recovery rates and relative standard deviations of the five nucleating agents (n=9)
2.6 实际样品检测
本次实验对多个工厂生产的20 余种不同牌号的PP 颗粒进行了成核剂检测,实际样品的第一次前处理统一采用MAE 温度80 ℃,时间10 min 预处理,得到的滤液用本实验优化得到的仪器方法,根据色谱峰保留时间确定实际样品中是否存在相关成核剂组分,再根据对应成核剂的最佳前处理条件,进行合适的前处理,最后通过外标曲线计算实际样品中成核剂含量。部分样品检测到成核剂相关成分,色谱出峰曲线如下图6所示,在3#、5#样中均检测到了NX8000,分别对峰面积进行积分计算,为326 874、408 748 μV·s,经外标曲线计算得实际添加量分别为1.2、1.5 g/kg;在2#中同时检测到了NA21和苯甲酸钠,峰面积积分分别为363 514、170 548 μV·s,经外标曲线计算得实际添加量分别为1.5、0.8 g/kg。

图6 实际样品色谱图Fig.6 Chromatogram of the actual samples
3 结论
(1)UPLC 条件为ACQUITY BEH C18(50 mm×2.1 mm,1.7 μm)柱,柱温箱35 ℃,流速0.3 mL/min,初始流动条件为30 %水相和70 %乙腈相,0~11 min内乙腈相线性升至99 %,11~15 min保持不变,每次进样量1 μL,PDA检测器波长为213 nm,实现5种成核剂在9 min内完全分离,精密度高,定量准确;
(2)比较了MAE、超声提取和加热回流提取方法的适用性,考察了前处理温度、时间、溶剂对提取效率的影响,发现MAE 提取效率高,重复性较好,溶剂用量少,是最佳的前处理方法;
(3)使用Design⁃Expert 软件以CCD 优化得到不同种类PP 成核剂的最佳MAE 条件,满足实际样品准确定量检测需要。
