LC-MS/MS测定舒必利原料药中基因毒性杂质(E)-1-乙基-2-(硝基亚甲基)吡咯烷
2023-01-16高雪闽邹巧根
高雪闽,邹巧根
(南京工业大学 药学院,江苏 南京 211800)
基因毒性杂质是指对DNA有潜在破坏性的,以及一些结构上类似基因毒性的物质,但尚未经实验证明[1]。基因毒性杂质存在于药物生产的各个阶段。原料药合成中的起始物料、中间体、副产物、催化剂和残留溶剂由于储存不当等原因,均可导致基因毒性杂质的产生[2]。基因毒性杂质会对人体产生极大的危害,目前各国药监部门对它们的质量控制越来越严格。
舒必利(sulpiride)是一种苯甲酰胺类药物,在临床上常作为联合用药来治疗精神类疾病,如:精神分裂、抑郁症等;也可用于胃病的治疗,如:消化不良和胃溃疡等;其化学结构见图1。王福兰等[3]发明了一种合成成本较低、纯度高且操作简便的生产路线,是用2-甲氧基-5-氨磺酰基苯甲酸甲酯(1)和N-乙基-2氨甲基四氢吡咯烷(2)缩合反应生成舒必利(图2)。物质2是以N-乙基吡咯烷酮(3)为原料,经Henry反应、加氢还原制得[3],合成路线见图3。图3中生成的中间体N-乙基-2-硝亚甲基吡咯烷(4)为芳香硝基类化合物,具有典型的基因毒性警示类结构[4]。在后续反应中,会通过混合溶剂进行提纯,以去除杂质,但由于舒必利的合成路线较短,所以4仍然有残留的可能。为保证舒必利的质量和用药安全,需对残留的4的反式异构体(E)-1-乙基-2-(硝基亚甲基)吡咯烷(5)进行严格控制,其中5的结构式见图4。
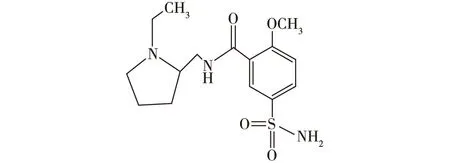
图1 舒必利的结构式Fig.1 The structure of sulpiride
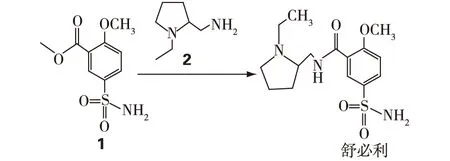
图2 舒必利的合成路线[3]Fig.2 The synthetic route of sulpiride[3]
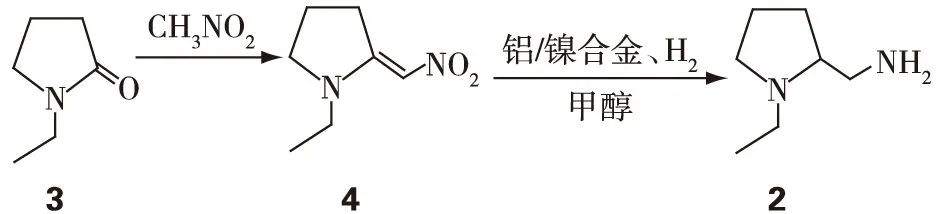
图3 2的合成路线[3-4]Fig.3 The synthetic route of 2[3-4]

图4 5的结构式Fig.4 The structure of 5
根据朱静等[5]的报道,5无诱变数据。对于已知具有致突变性但致癌性未知的杂质,一般采用毒理学关注阈(TTC)来评估其毒性[5-8],即通常认为:人体每日摄入量低于1.5 μg,可诱发癌变的物质是可以接受的。舒必利原料的最大日摄入剂量为1.2 g,按照TTC不高于1.5 μg进行控制,则杂质限度应控制在1.25 μg/g[9]。
基因毒性杂质都是以痕量的形式存在于药物中,因此选择合适的分离及检测方法尤为重要。液相色谱-串联质谱法(LC-MS/MS)由于具有高选择性和高灵敏度,是近年来检测基因毒性杂质常用的技术[10-13]。目前尚无文献对5的测定方法进行报道。本文首次建立了一种快速灵敏的测定舒必利原料药中5的LC-MS/MS分析方法,且对此方法进行了全方位的验证,以期将其成功应用于舒必利原料药中基因毒性杂质5的分析。
1 材料与方法
1.1 仪器与试剂
Agilent1100-API4000Q型LC-MS/MS,美国Agilent-Applied Biosystems Sciex公司;XS105型十万分之一天平、XP6型百万分之一天平,瑞士Mettled Toledo公司;Direct-Q3UV型纯水仪,上海市密理博有限公司。
5对照品(批号:PAS-120-14),含量99.15%,美国AOCS公司;舒必利原料药(批号:S2003002、S2003003和S2003004),自制;甲醇、乙腈为色谱纯,美国Merck公司;甲酸为市售色谱纯,上海市Aladdia公司。
1.2 实验方法
1.2.1 色谱条件
色谱柱为ZORBAX Eclipse XDB-Phenyl型(4.60 mm×150 mm,5.0 μm);柱温为30 ℃;流动相A为甲醇;流动相B为0.01%(体积分数,下同)甲酸水溶液;流速为0.8 mL/min;分析时间为11 min;进样量为5 μL。
梯度洗脱程序为0~0.1 min,70% B;0.1~5 min,70% B;5~8 min,70%~60% B;8~9.5 min,60% B;9.5~9.7 min,60%~70% B;9.7~11 min,70% B。
1.2.2 质谱条件
电喷雾离子源(ESI);正离子模式,多重反应检测模式;离子源温度为550 ℃,离子源电压为5 500 V,检测离子对为97.4~157.4 质荷比(m/z),去簇电压和碰撞能量分别为40 和27 V;二级质谱见图5。
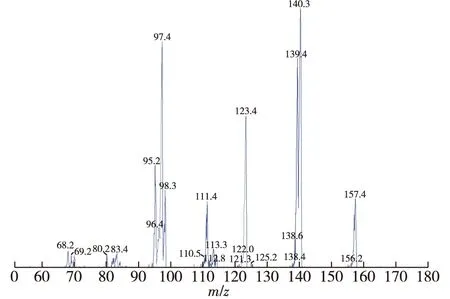
图5 5在正离子模式下的离子源扫描Fig.5 The product ion scans chromatogram of 5 in the positive mode
1.2.3 对照品一级储备液的制备
精密称定5对照品5 mg至10 mL容量瓶中,用50%乙腈水溶液(含0.1%甲酸)溶解,并定容至刻度线,摇匀。
1.2.4 对照品二级储备液的制备
精密量取对照品一级储备液1 mL,用50%乙腈水溶液(含0.1%甲酸)稀释,得到质量浓度为50 ng/mL的对照品二级储备液。
1.2.5 对照品溶液的制备
精密量取对照品二级储备液适量,用50%乙腈水溶液(含0.1%甲酸)稀释,得到质量浓度为25 ng/mL的对照品溶液。
1.2.6 供试品溶液的制备
精密称取原料药100 mg,加入50%乙腈水溶液(含0.1%甲酸)5 mL,得到终质量浓度为20 mg/mL的供试品溶液。
1.2.7 加标供试品溶液的制备
精密称取原料药100 mg,加入对照品二级储备液2.5 mL,用50%乙腈水溶液(含0.1%甲酸)溶解,得到含质量浓度为25 ng/mL的对照品溶液和舒必利质量浓度为20 mg/mL的加标供试品溶液。
1.2.8 线性系列标准溶液的制备
分别取对照品二级储备液适量,用50%乙腈水溶液(含0.1%甲酸)稀释成相当于限度水平30%、50%、80%、100%与120%的系列浓度溶液。
1.2.9 回收率试验溶液的制备
精密称取原料药100 mg,分别加入对照品二级储备液0.75、2.5、3 mL,用50%乙腈水溶液(含0.1%甲酸)溶解,每个浓度分别制备3份,分别得到30%、100%、120%限度水平的回收率试验溶液。
1.2.10 定量限与检测限溶液的制备
取对照品溶液逐级稀释,分别以信噪比(S/N)为10和3时的溶液作为定量限溶液与检测限溶液。
1.2.11 试剂空白溶液的制备
配制50%乙腈水溶液(含0.1%甲酸)。
1.2.12 分析方法的验证
参考文献[14-15]的方法,对所建立的分析方法进行验证,验证项目包括专属性、线性范围、检测限、定量限、准确度、精密度(包括重复性、进样精密度和中间精密度)、稳定性和耐用性。
2 结果与讨论
2.1 专属性试验结果
取空白溶液、供试品溶液、对照品溶液及加标供试品溶液分别进样测定,其专属性样品色谱流出曲线的结果见图6。由图6可知:杂质的保留时间为6.87 min,空白溶液及供试品溶液都不会干扰杂质的检测,杂质与主成分之间也互不干扰,因此表明本方法的专属性良好。
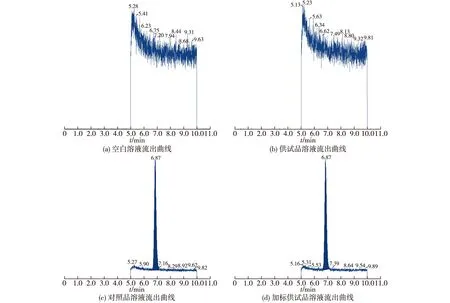
图6 专属性样品色谱流出曲线Fig.6 Chromatographic elution curves of specific samples
2.2 线性试验结果
以进样质量浓度(ng/mL)为横坐标(X),峰面积为纵坐标(Y),进行线性回归分析,得到的线性方程为Y=8 580X+3 950,R2=0.998 4。表明5在7.5~30 ng/mL范围内均呈现出良好线性水平,由此说明:浓度范围包含杂质限度所要求的30%~120%,可以满足分析要求。
2.3 定量限与检测限试验结果
取定量限与检测限溶液进样测定。将定量限溶液重复测定6次,得到峰面积的相对标准偏差(RSD)为3.8%,定量限质量浓度为3.00 ng/mL,检测限质量浓度为1.50 ng/mL。结果表明本方法的灵敏度高。
2.4 回收率试验结果
取空白溶液和回收率试验溶液进样分析。计算5测定值与理论值的比值,以获得回收率,结果见表1。由表1可知:检测物的平均回收率分别为98.09%、94.27%和97.78%,总体回收率为96.71%,RSD为2.40%。以上结果表明本方法的准确度良好。

表1 回收率试验结果
2.5 精密度试验结果
2.5.1 进样精密度的试验结果
取对照品溶液连续进样6针,考察进样的精密度,结果见表2。由表2可知:对照品溶液中5的批间保留时间RSD为0.20%,批内保留时间RSD为2.20%,均符合验证要求。表明本方法的进样精密度良好。
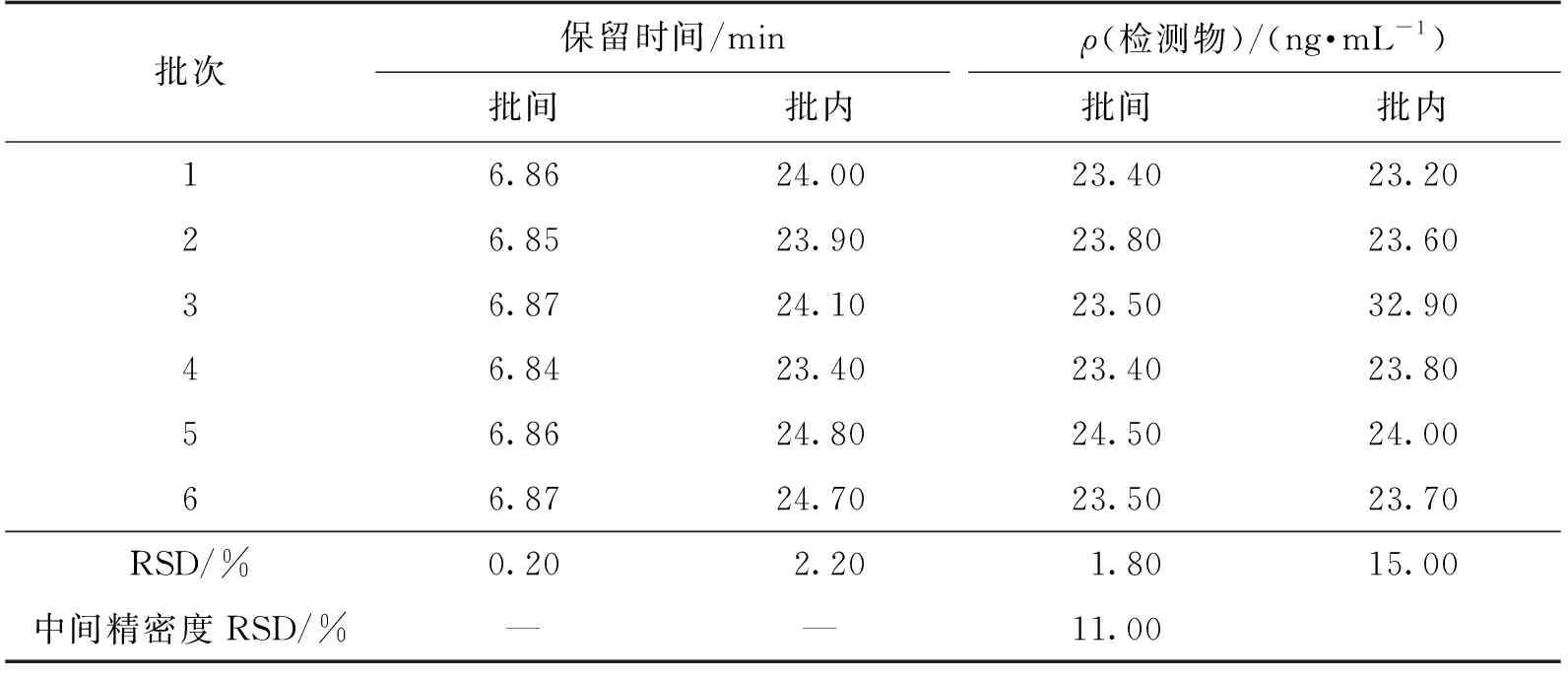
表2 精密度试验结果
2.5.2 重复性的试验结果
取加标供试品溶液,平行配制6份进行进样分析,结果见表2。由表2可知:检测物5的批间RSD为1.80%,表明本方法重复性良好。
2.5.3 中间精密度的试验结果
取加标供试品溶液,由不同分析人员在不同时间再次制备6份加标供试品溶液进样测定,结果见表2。由表2可知:检测物5的批间中间精密度RSD为11.00%。表明本方法的中间精密度良好。
2.6 稳定性试验结果
取对照品溶液和供试品溶液,分别在室温条件下放置0、2.0、4.8、7.7、11.0、14.4、17.7和21.0 h之后进样测定,检测结果见表3。由表3可知:对照品溶液室温放置21.0 h后,5的峰面积RSD为5.80%,无明显变化;供试品溶液室温放置21.0 h后,均未有5检出。表明对照品溶液和供试品溶液于室温放置21.0 h后均能保持稳定。
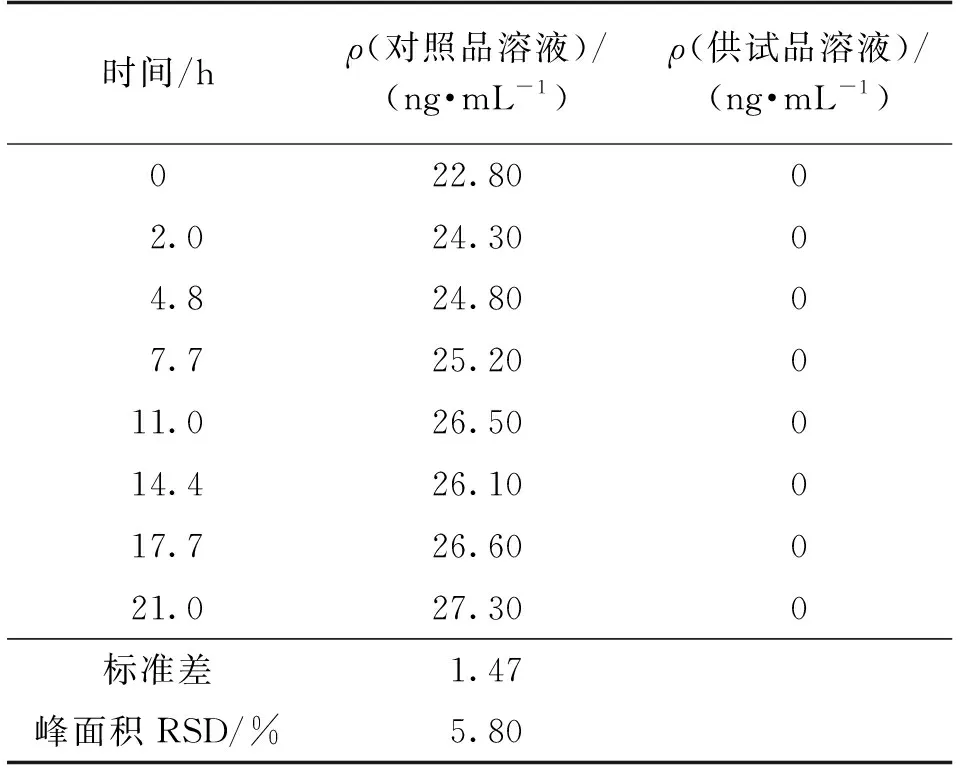
表3 溶液稳定性试验结果
2.7 耐用性试验结果
取加标供试品溶液进样分析,考察其在柱温为(30±5) ℃、流速为(800±50) μL/min以及同品牌不同批号的色谱柱条件下的耐用性,结果见表4。由表4可知:当柱温为(30±5) ℃,流速为(800±50)μL/min,色谱柱发生微小变动时,RSD分别为0.7%、3.2%和2.1%,均符合验证要求。表明本方法在发生微小变动时,耐用性仍保持良好。
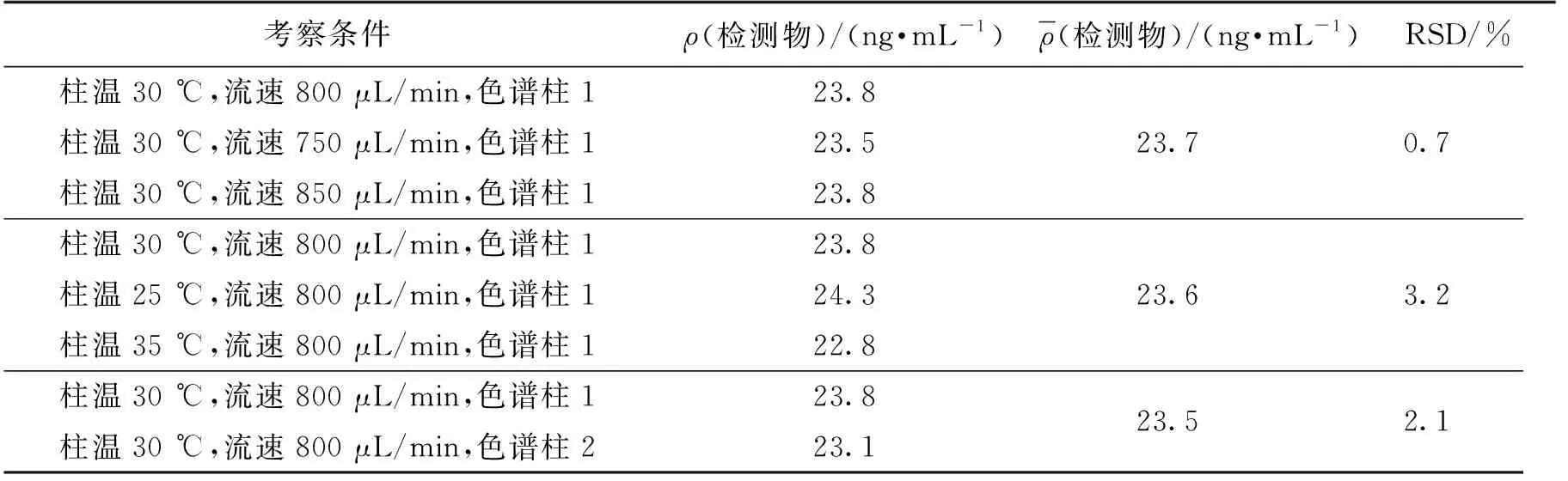
表4 耐用性试验结果
2.8 样品测定结果
采用经过验证的方法,测定3个批次原料药样品中5的含量,每批次分别进行2次重复试验。按供试品溶液进行处理后的进样分析,测定结果见表5。由表5可知:3个批次中均未检出5。

表5 3批舒必利原料药中5的检测结果
3 结论
本文建立了舒必利原料药中(E)-1-乙基-2-(硝基亚甲基)吡咯烷的LC-MS/MS检测方法。该方法的专属性强、灵敏度高、溶液稳定性好,可以满足杂质定量检测要求,可作为舒必利原料药中检测(E)-1-乙基-2-(硝基亚甲基)吡咯烷的研究方法。该方法不仅完善了舒必利的质量控制体系,并为其他类药物中涉及到N-乙基-2-硝亚甲基吡咯烷类基因毒性杂质的质量控制提供了理论基础和技术手段。
