扎冲十三味丸HPLC指纹图谱研究
2023-01-12王会品王金梅刘丹娜
王会品,王金梅,刘丹娜
1郑州市第三人民医院,郑州 450000;2河南大学药学院,郑州 475000
扎冲十三味丸由诃子、制草乌、木香、石菖蒲、珊瑚(制)、人工麝香、丁香、珍珠(制)、沉香、肉豆蔻、磁石(煅)、禹粮土、甘草共13味药材制得,具有祛风通窍、舒筋活血、镇静安神功效,多用于半身不遂、口眼歪斜、腰腿不利、四肢麻木、筋骨疼痛、言语不清、神经麻痹、风湿及关节疼痛等[1]。方中诃子被誉为“藏药之王”,具有抗氧化、抗病毒、抗肿瘤、抗菌和神经保护等多种药理作用,内含活性成份没食子酸、槲皮素和鞣花酸等[2-4]。石菖蒲的药理活性成份为挥发油α细辛醚、β细辛醚和细辛醛等成份,多用于神昏癫痫、健忘失眠、耳鸣耳聋、脘痞不饥和噤口下痢[5-6]。甘草在成方制剂中应用广泛,素有“十方九草”之称,其含有的三萜类成份中以甘草酸含量最高,具有抗炎、免疫调节及抗肝损伤等生物活性,甘草含有的黄酮类成份主要有甘草素、甘草苷等,具有抑菌、抗炎、抗病毒及抗氧化等作用[7-9]。木香中含有丰富的萜类成份,主要有抗炎、抗肿瘤和抗溃疡的作用,代表性成份有木香烃内酯和去氢木香内酯[10-11]。扎冲十三味丸收载于《中华人民共和国卫生部药品标准》蒙药分册[1]中,质量标准项下未对其内在活性提出定量质控要求,相关文献[12-14]也只是对其含有的个别成份进行考察,未见有关指纹图谱方面报道。
中药指纹图谱能客观合理、科学全面地评价中药质量,在中药质量分析领域应用广泛[15-16]。本研究收集了2个不同厂家、共14批次的扎冲十三味丸样品,建立HPLC指纹图谱,并对指纹图谱进行聚类分析及主成份分析,考察不同产地扎冲十三味丸的内部质量,为系统客观、科学评价扎冲十三味丸质量提供参考依据。
1 材料
1.1 仪器
2695型高效液相色谱仪(DAD检测器,美国Waters公司);AL204型分析天平(十万分之一,瑞士Mettler Toledo公司);SDPP制样粉碎机(湖南三德科技股份有限公司);KQ-600DB型超声波清洗器(昆山超声波仪器有限公司)。
1.2 试药
α细辛醚对照品(成都瑞芬思生物科技有限公司,批号X-037-150921,含量≥99.0%);木香烃内酯对照品(批号:111524-201911,含量99.9%)、鞣花酸对照品(批号111959-201602,含量89.3%)、没食子酸对照品(批号110831-201906,含量91.5%)、去氢木香内酯对照品(批号111525-201912,含量99.5%)、槲皮素对照品(批号100081-201610,含量99.1%)、甘草苷对照品(批号111610-201607,含量93.1%)、甘草酸铵对照品(批号110731-202021,含量96.2%)均购自中国食品药品检定研究院;乙腈(美国天地公司,色谱纯);水为娃哈哈纯净水;其余试剂均为分析纯。
实验用14批扎冲十三味丸的规格均为2g/10粒。其中,S1~S8由阜新蒙药有限责任公司生产,批号分别为:20170901、20170902、20180302、20180405、20180502、20180507、20180601、20180905;S9~S14由内蒙古大唐药业股份有限公司生产,批号分别为:170203、170512、170605、180401、180502、181005。
2 方法与结果[13]
2.1 色谱条件
Waters Tnature C18色谱柱(250mm×4.6mm,5μm);流动相为乙腈-0.1%磷酸溶液,梯度洗脱(0~5min,5%乙腈;5~18min,5%→12%乙腈;18~25min,12%→28%乙腈;25~35min,28%→40%乙腈;35~40min,40%乙腈;40~55min,40%→58%乙腈;55~75min,58%乙腈;75~80min,58%→5%乙腈);检测波长为254nm;流速为1.0ml/min;柱温为36℃;进样量为10μl。
2.2 溶液的制备
2.2.1 混合对照品溶液的制备
取本研究指认共有峰所需对照品各适量,用甲醇溶解后配制成每1ml含没食子酸0.125mg、鞣花酸0.321mg、甘草苷0.209mg、槲皮素0.225mg、甘草酸铵0.189mg、木香烃内酯0.211mg、去氢木香内酯0.165mg、α细辛醚0.351mg的混合对照品储备溶液。精密量取混合对照品储备液5ml,置25ml量瓶中,加甲醇稀释至刻度,摇匀,用0.45μm微孔滤膜滤过,取续滤液,即得混合对照品溶液。
2.2.2 供试品溶液的制备
取扎冲十三味丸,粉碎,过80目筛。取细粉约2.0g,精密称定,置具塞锥形瓶中,精密加入50%甲醇50ml,闭塞,称定重量,超声处理(功率250W,频率60kHz)1h,放冷,再称定质量,用50%甲醇溶液补足减失重量,摇匀,用0.45μm微孔滤膜滤过,取续滤液,即得。
2.3 方法学考察
2.3.1 精密度试验
取扎冲十三味丸供试品溶液(批号170203),按“2.1”项下所述色谱条件连续进样6次,记录共有峰色谱图,以槲皮素为参比峰,计算其他24个共有峰的相对保留时间和相对峰面积。结果显示,各共有峰相对保留时间的RSD为0.2%~0.9%,各共有峰相对峰面积的RSD为0.5%~1.2%,表明仪器精密度良好。
2.3.2 稳定性试验
取同一批扎冲十三味丸供试品溶液(批号170203),在室温放置0、3、6、9、12、18、24h后,按“2.1”项下所述色谱条件测定,记录各共有峰色谱图,以槲皮素为参比峰,计算其他24个共有峰的相对保留时间和相对峰面积。结果显示,各共有峰相对保留时间的RSD为0.5%~1.2%,各共有峰相对峰面积的RSD为1.0%~2.0%,表明扎冲十三味丸供试品溶液在24h内稳定性良好。
2.3.3 重复性试验
取同一批扎冲十三味丸供试品(批号170203)6份,按“2.2.2”项下方法制备供试品溶液,按“2.1”项下所述色谱条件测定,记录各共有峰色谱图,以槲皮素为参比峰,计算其他24个共有峰的相对保留时间和相对峰面积。结果显示,各共有峰相对保留时间的RSD为0.4%~1.1%,各共有峰相对峰面积的RSD为0.7%~1.5%,表明本方法重复性良好。
2.4 扎冲十三味丸HPLC指纹图谱的建立及分析
2.4.1 指纹图谱的建立
取14批扎冲十三味丸样品,按“2.2.2”项下方法制备供试品溶液,按“2.1”项下所述色谱条件测定,记录各批样品色谱图。将14批扎冲十三味丸供试品色谱图在同条件下积分处理后,导出AIA格式数据至“中药色谱指纹图谱相似度评价系统(2012版)”软件中,设定扎冲十三味丸S1号样品色谱图为参考图谱,设定时间窗宽度为0.2min,采用多点校正法对14批样品色谱图进行自动匹配分析,得到扎冲十三味丸对照指纹图谱(见图1)和14批扎冲十三味丸HPLC指纹图谱叠加图谱(见图2),共确定24个共有峰。

图1 扎冲十三味丸对照指纹图谱

图2 14批扎冲十三味丸样品HPLC叠加图谱(S1~S14)
2.4.2 相似度评价分析
设定S1号供试品色谱图为参考图谱,对14批扎冲十三味丸指纹图谱进行整体相似度评价。结果,14批扎冲十三味丸供试品指纹图谱相似度为0.960~0.993,说明不同生产厂家扎冲十三味丸指纹图谱较为相似。相似度评价数据见表1。

表1 14批扎冲十三味丸样品相似度评价数据
2.4.3 共有峰指认及相对峰面积计算
通过与混合对照品HPLC色谱图(见图3)比对,24个共有峰中共指认出8个成份,即峰5为没食子酸、峰10为鞣花酸、峰13为甘草苷、峰15为槲皮素、峰20为甘草酸铵、峰21为木香烃内酯、峰22为去氢木香内酯、峰24为α细辛醚。其中,以峰15(槲皮素)的峰面积较大,位置适中,故设定为参照峰,计算其余23个色谱峰相对峰面积,见表2。

图3 混合对照品HPLC色谱图
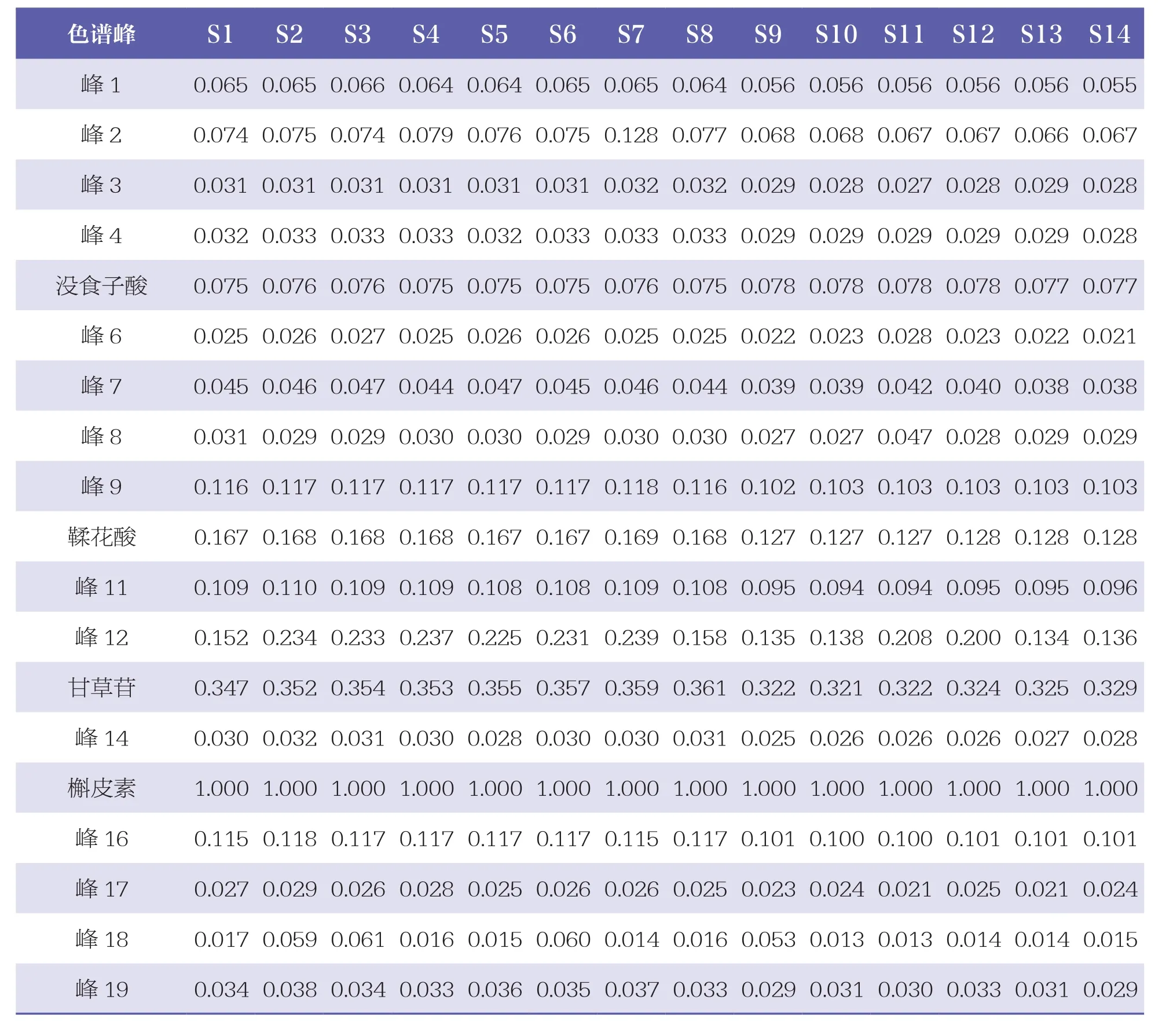
表2 14批扎冲十三味丸样品HPLC图谱共有峰的相对峰面积
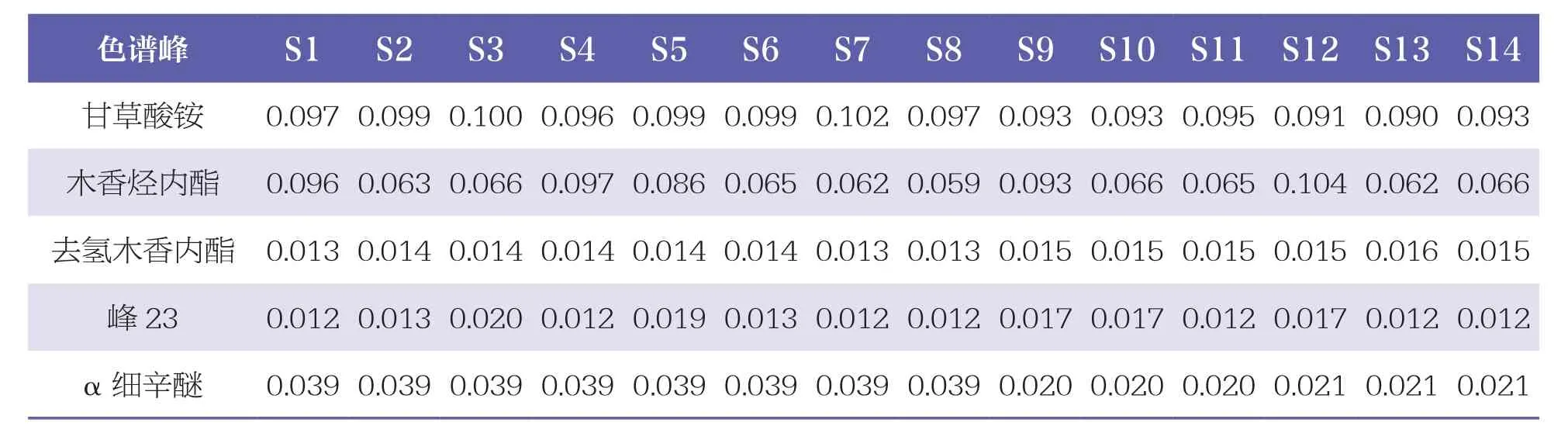
续表
2.5 聚类分析
为考察14批扎冲十三味丸样品间差异,本研究采用SPSS 22.0软件,以14批扎冲十三味丸指纹图谱峰面积为变量,采用平均欧式距离法对样本进行聚类分析,结果见图4。由图可知,14批扎冲十三味丸大致分为2类,Ⅰ类包括S1、S2、S3、S4、S5、S6、S7、S8,Ⅱ类包括 S9、S10、S11、S12、S13、S14。结合样品信息可知,Ⅰ类均为阜新蒙药有限责任公司生产,Ⅱ类为内蒙古大唐药业股份有限公司生产。可见,不同厂家扎冲十三味丸之间相关性和相似度结果较为一致。

图4 14批扎冲十三味丸样品聚类分析树状图
2.6 主成份分析
以扎冲十三味丸指纹图谱中24个共有峰面积为变量,利用SPSS 22.0软件,共提取特征值大于1的主成份3个,累计方差贡献率为90.162%。其中,3个主成份特征值分别为19.072、1.482、1.084,其方差贡献率分别为79.467%、6.176%、4.519%,且前3个主成份累计方差贡献率为90.162%,提示模型预测性良好,见表3。初始因子载荷矩阵结果显示(见表4),主成份1主要体现峰1、峰3、峰4、没食子酸、峰6、峰7、峰9、鞣花酸、峰11、甘草苷、峰14、槲皮素、峰16、峰17、峰19、甘草酸铵及α细辛醚的信息。
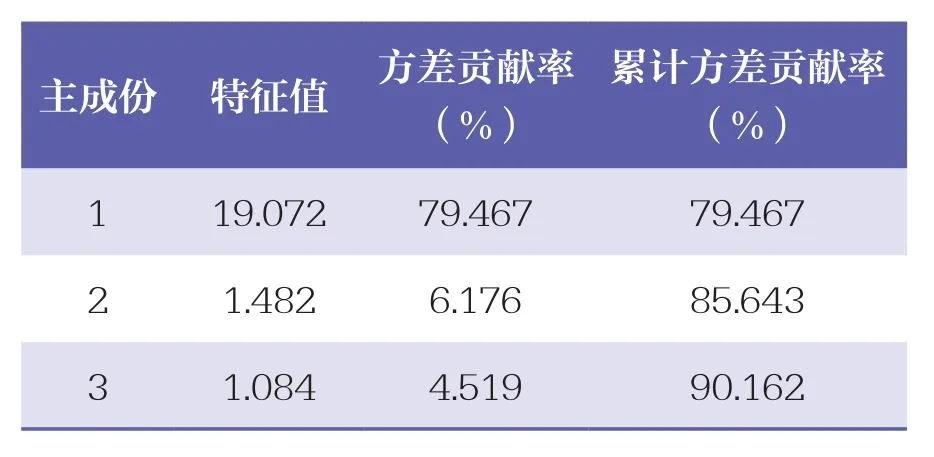
表3 3个主成份的特征值及方差贡献率

表4 初始因子载荷矩阵

续表
用SIMCA 14.1分析软件对14批样品共有峰面积进行主成份分析,主成份得分图见图4。结果表明,14批扎冲十三味丸样品被分为2类,与“2.5”项下聚类分析结果一致。

图4 14批样品主成份分析得分图
3 讨论
3.1 色谱条件的优化
结合文献[13-14],在进行色谱条件优化过程中,以乙腈-0.1%磷酸水溶液作为流动相时,指纹图谱峰质量较好,各目标成份均能有效分离,且信号强度较好。波长选取时,在190~400nm波段分别对没食子酸、鞣花酸、甘草苷、槲皮素、甘草酸铵、木香烃内酯、去氢木香内酯和α细辛醚对照溶液进行全波长扫描,发现在254nm处各成份均有较强吸收,且次波长处指纹图谱中色谱峰个数较多,所以本研究采用254nm为监测波长。
3.2 提取条件的优化
扎冲十三味丸由13味药材制得,含有多种成份,因不同组份存在极性差异,所以样品有效成份的提取步骤对实验开展十分重要。为探索供试品制备方法,本实验采用L9(34)正交试验法,选择甲醇浓度、溶剂用量、超声提取时间为3个因素,以指纹图谱的峰面积之和为指标进行考察,结果以每2g成药粉末加50%甲醇,超声提取1h效果最佳。
3.3 指纹图谱分析
本研究从14批样品的指纹图谱中共提取到24个共有峰,通过与混合对照品色谱图比对,指认了其中8个成份。为进一步考察扎冲十三味丸的组内和组间质量,对14批样品进一步进行聚类分析和主成份分析,结果14批样品聚为2类,说明不同厂家扎冲十三味丸可能因处方、工艺等因素不同而导致指纹图谱存在差异,但同一厂家均匀性较好。在进行主成份分析时,从24个共有峰中共提取到3个主成份,累计方差贡献率达90.162%,提示其能充分反映指纹图谱绝大部分信息,且指认的8个成份在3个主成份中均有较大贡献率,说明8个成份代表性较佳,可作为药品的质控指标,用于客观合理评价扎冲十三味丸的质量。
4 小结
中药及其制剂均为多组份复杂体系,因此评价其质量应采用与之相适应的、能提供丰富鉴别信息的检测方法,但现行的显微鉴别、理化鉴别和含量测定等方法都不足以解决这一问题。建立中药指纹图谱能较为全面地反映中药及其制剂中所含化学成份的种类与数量,进而对药品质量进行整体描述和评价,这也正好符合中医药理论下的整体学说。扎冲十三味丸由13味药材制成,简单的多成份分析方式代表性欠佳,而指纹图谱可客观、系统地对扎冲十三味丸进行质量评价,有利于科学监管药品的质量。本实验首次建立了扎冲十三味丸HPLC指纹图谱法,方法简单、实用,方法学考察专属性强,可有效评价扎冲十三味丸质量,为进一步完善扎冲十三味丸的标准提供了一定的实验依据。
