过渡金属(Mo,Ru,Rh,Pd)掺杂SnO2 磁学性质的第一性原理研究
2023-01-10雷博程刘晨曦张丽丽赵旭才黄以能
雷博程,刘晨曦,张丽丽,赵旭才,黄以能,2
(1.伊犁师范大学 物理科学与技术学院 新疆凝聚态相变与微结构重点实验室,新疆 伊宁 835000;2.南京大学 物理学院 固体微结构物理国家重点实验室,江苏 南京 210093)
近年来,自旋电子学研究领域在不断拓宽,现已成为集材料科学、信息技术和凝聚态物理等多门学科于一体的新型学科[1]。研究人员发现将磁性溶于半导体材料中能使其同时具有电子的电荷和自旋属性,得到一类低能耗、高速处理和对信息“非易失性” 的新器件——稀磁半导体(Diluted Magnetic Semiconductor,DMS)[2-4]。自20 世纪60-70 年代以来,研究者们开始利用过渡金属掺杂单质半导体、II-VI 族或III-V 族化合物等,发现能制成DMS 的基底材料不胜枚举[5-6],其中SnO2因具有独特的电学、磁学以及稳定的物理化学特性,成为了众多基底材料中最具代表性的金属氧化物半导体之一[7-8]。然而,本征SnO2是宽禁带半导体,在理论上是绝缘态的(电阻率过大),且室温下没有铁磁性产生,这极大地限制了SnO2在自旋电子器件、半导体集成电路以及磁感应器等领域的应用。为了解决上述问题,研究者们尝试采用掺杂的方式使SnO2具有DMS 的特性,如在实验上制备了Li[9]、V[10]、Fe[11]、Co[12]或Ni[13]元素掺杂SnO2,可发现在室温下有铁磁性产生。Ogale 等[14]运用脉冲激光沉积技术制备了Co 掺杂SnO2薄膜,其铁磁性可保持在较高居里温度,Co 离子的平均磁矩高达(7.5±0.5)玻尔磁子(μB)。相似的,Stoian 等[15]也在Co 掺杂SnO2薄膜中检测出室温铁磁性。除此之外,还有一些研究组采用第一性原理的方法对金属元素(Li、K、Be、Mg、Ca[16]、Na[17]、Ga[18]) 掺杂SnO2和过渡金属元素(Ag[19]、Fe、V、Cr[20]、Mn[20-21])掺杂SnO2体系进行了研究,发现掺杂体系均出现了室温铁磁性和半金属特性。但是,在过渡金属掺杂SnO2这个研究领域仍然存在很多争议性的问题,比如磁性的起源还没有一个统一的定论,并且当掺杂磁性过渡金属元素时,因来自磁团簇或次级相的铁磁性不易制成磁性半导体器件。有大量的实验和理论工作表明,避免磁性沉淀有关问题的可行方法是选择用非磁性过渡金属元素掺杂氧化物半导体[22-23]。此外,若材料的居里温度远低于室温则实际应用困难,达不到工业化水平,因此如何获得居里温度高于室温的DMS 材料也是亟待解决的问题之一。
本文用第一性原理的自旋极化密度泛函理论方法,构建了非磁性过渡金属元素(Mo,Ru,Rh,Pd)对SnO2进行掺杂,研究掺杂后是否产生室温铁磁性,进一步探讨SnO2-DMS 材料铁磁性的起源和物理机制是否如预估的一样来源于非磁性元素的掺杂,期望能对相关实验的研究提供理论依据和参考,以更好地运用到未来的自旋电子器件中。
1 理论模型与计算方法
1.1 模型构建
金红石结构的SnO2属于四方晶系,其空间群为P42/MNM(No.136),晶格常数分别为a=b=0.4830 nm,c=0.3236 nm;α=β=γ=90°[24],原子坐标分别为Sn(0,0,0),O(0.306,0.306,0),其中Sn4+位于晶胞顶角和体心位置,其配位数为6,周围有6 个O2-,构成氧八面体;为了减小边界效应,过渡金属将替代体心位置的Sn 原子,共建立4 种[(2×2×2)、(2×2×3)、(2×3×3)、(3×3×3)] 模型,分别为Sn0.9375X0.0625O2、Sn0.9583X0.0417O2、Sn0.9722X0.0278O2及Sn0.9815X0.0185O2(X 为Mo/Ru/Rh/Pd),如图1(a)~(d)所示。

图1 (a)Sn0.9375X0.0625O2、(b)Sn0.9583X0.0417O2、(c)Sn0.9722X0.0278O2和(d)Sn0.9815X0.0185O2(X=Mo/Ru/Rh/Pd)的结构模型Fig.1 Structural models of (a)Sn0.9375X0.0625O2,(b)Sn0.9583X0.0417O2,(c)Sn0.9722X0.0278O2 and(d)Sn0.9815X0.0185O2(X=Mo/Ru/Rh/Pd)
各原子的价电子组态分别为: Sn(5s25p2),O(2s22p4),Mo(4d55s1),Ru(4d75s1),Rh(4d85s1),Pd(4d10),其中过渡金属掺杂的原子比例分别为6.25%,4.17%,2.78%和1.85%。虽然这些超胞对于模拟真实的实验条件来说不是很大,但它们在计算模拟中,满足周期性对称条件,超胞就足够大,可以模拟X(Mo/Ru/Rh/Pd)掺杂剂的代表性浓度的主要性质。
1.2 计算方法
本文基于密度泛函理论的第一性原理,利用Materials Studio 中的CASTEP[25]模块完成计算。对交换关联函数采用局域密度近似(Local Density Approximation,LDA)[26]下的CA-PZ 进行计算,能量计算的收敛精度为2×10-6eV/atom;内应力小于0.1 GPa;公差偏移量为0.0002 nm,单原子受力小于0.5 eV/nm。因为合适的平面波截断能(Ecut)不仅可以获得准确的计算结果,还能节约计算资源,因此本文对截断能进行了收敛性测试(如图2),Ecut取为420 eV;根据Monkhorst-Pack 方案优化时的K 点选取为2×2×2,所有计算均在倒易空间进行。
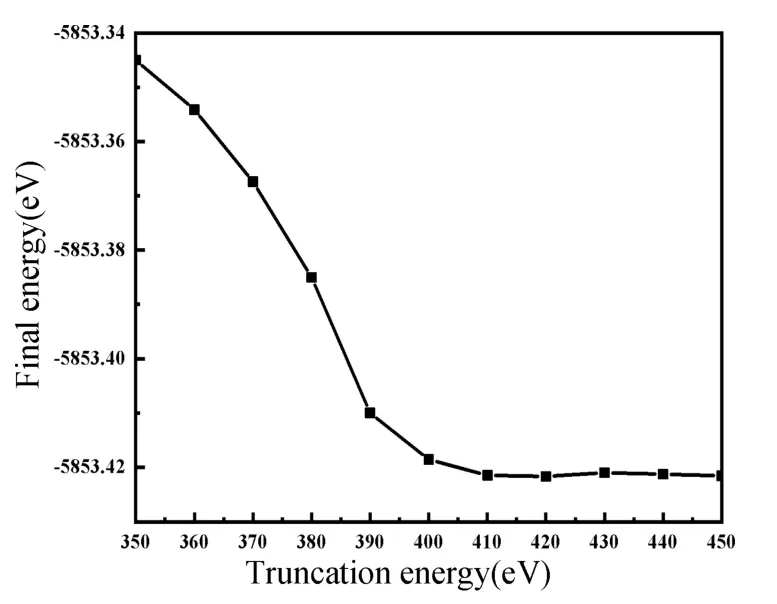
图2 截断能收敛性测试Fig.2 Convergence test of truncation energy
其中,对于以上四种掺杂模型,选取了总能量最低、磁性最好且满足周期性边界条件的模型(2×2×2),再进行三种自旋态设置(无自旋(NM)/铁磁(FM)/反铁磁(AFM))。即通过软件中单原子自旋设置功能,按晶面切分,依次将每个晶面原子的自旋方向设置为无自旋/同向/反向[27-28],再计算每个自旋方式的基态总能量,用来确定各掺杂模型的磁性状态,为后文的计算提供稳定的基底(如表1)。
2 计算结果及讨论
2.1 结构优化与稳定性分析
表1 给出了SnO2计算前后各模型的物理参数。从表1 可以看出,将优化后纯SnO2与实验值[24]做了误差分析,晶格常数a(b)的误差为1.192%,c的误差为1.081%,a/c的误差为0.113%,体积的误差为3.457%;而掺入过渡金属(Mo,Ru,Rh,Pd)后,与纯SnO2相比,晶格常数a(b)、c、a/c和V的误差都在0.1%~0.6%之间,说明计算后的晶格常数误差很小,故采用的计算方法及参数设置合理、可靠。还可看出,Mo、Ru 和Rh 掺杂SnO2体系时FM 态的基态总能量最低,Pd 掺杂时NM 态的基态总能量最低(如表1),考虑到体系配置的基态能量越低越稳定,所以本文对于Mo/Ru/Rh 掺杂SnO2体系时取铁磁态配置,Pd 掺杂取无磁态配置,下文将进一步验证掺杂体系的磁性状态。
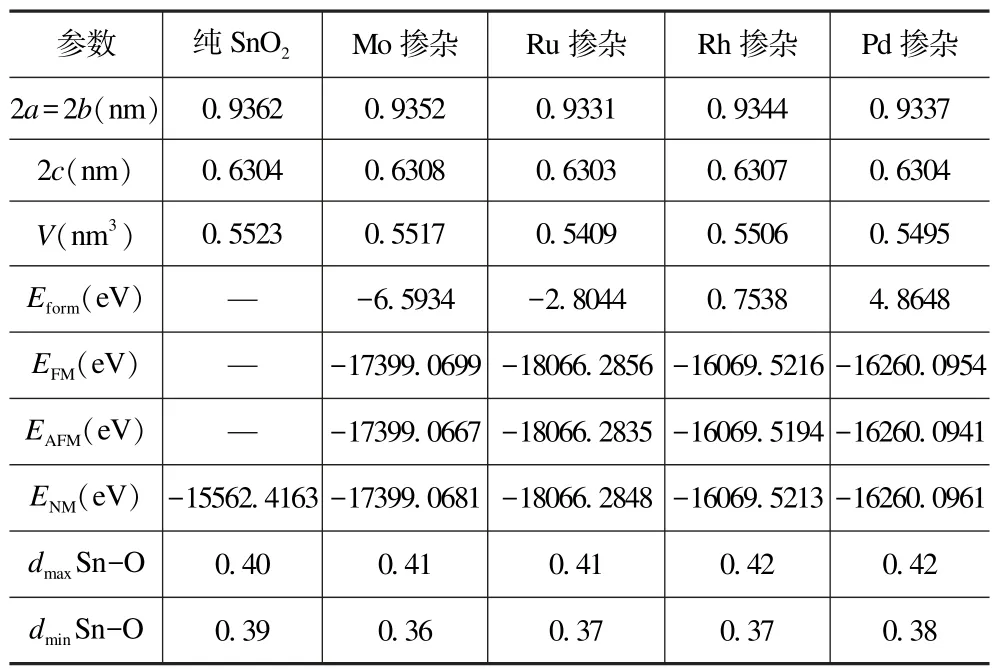
表1 所有计算模型的物理参数Tab.1 Physical parameters of all compute models
为了反映杂质在基体材料中形成的难易程度,因此计算了Sn0.9375X0.0625O2(X=Mo/Ru/Rh/Pd)体系的形成能。其计算公式如下[29]:

式中:Eform是掺杂形成能;Edoped是缺陷晶体的总能量;Eperfect是完美晶体的基态能量;μX和μSn是过渡金属元素和Sn 元素的化学势;n、m分别为掺入的过渡元素和被替换的Sn 元素个数(其中n=m=1)。式(1)反映了掺杂前后体系的能量差,Eform越大,表明杂质越难掺入基体材料,Eform越小,表明杂质越易掺入基体材料。由表1 中Eform计算结果可知,随着掺杂原子半径的增大,形成能越来越小,说明杂质越来越容易掺入基体材料;其中Sn0.9375Mo0.0625O2体系的形成能最小,相比于Ru,Rh 和Pd 掺杂SnO2,Mo 掺杂SnO2最易生成,并且溶解度最高,容易获得高浓度的Mo 掺杂SnO2。而掺入原子Rh 和Pd 后,形成能变为正值,表明杂质难掺入基体材料,因为Rh 和Pd 的电子组态分别为Rh(4d85s1)和Pd(4d10),在d 轨道趋于饱和,化学性质不活泼,当掺入基体材料时,不易分离电子,与基体材料不结合。
根据布局数的概念[30],键布局数为正,说明原子之间是成键的,且高值表示键为共价键,低值表示原子之间是离子相互作用;键布局数为负,说明原子之间是反键,即不成键。掺入过渡金属元素后,各体系的最大与最小Sn—O 键布局数随原子电荷数的增加而增大,其共价性也有相同的变化趋势,说明Sn—O 的共价性的强弱与掺入原子的序数成正比,且与形成能结果一致,表明计算结果合理。
2.2 磁性机理
态密度的分布是判断磁性机理的一个重要因素,因此计算了掺杂前后的电子态密度图,设置了能量0 eV 为费米能级。由于-10 eV 以下和15 eV 以上的能级距离费米能级较远,与导带和价带其他部分相互作用较弱,且对费米能级附近电子态作用影响不大,所以没有对此部分进行讨论。
如图3(a)所示,金红石相SnO2的导带与价带均由Sn-5p 态与O-2p 态杂化构成,并且各个自旋向上和向下电子密度的峰值都十分接近,其中Sn-5p 态对导带起主要作用,O-2p 态对价带起主要作用,自旋向上和向下的态密度分布完全对称,费米能级附近未产生自旋劈裂,表明纯SnO2无磁性。
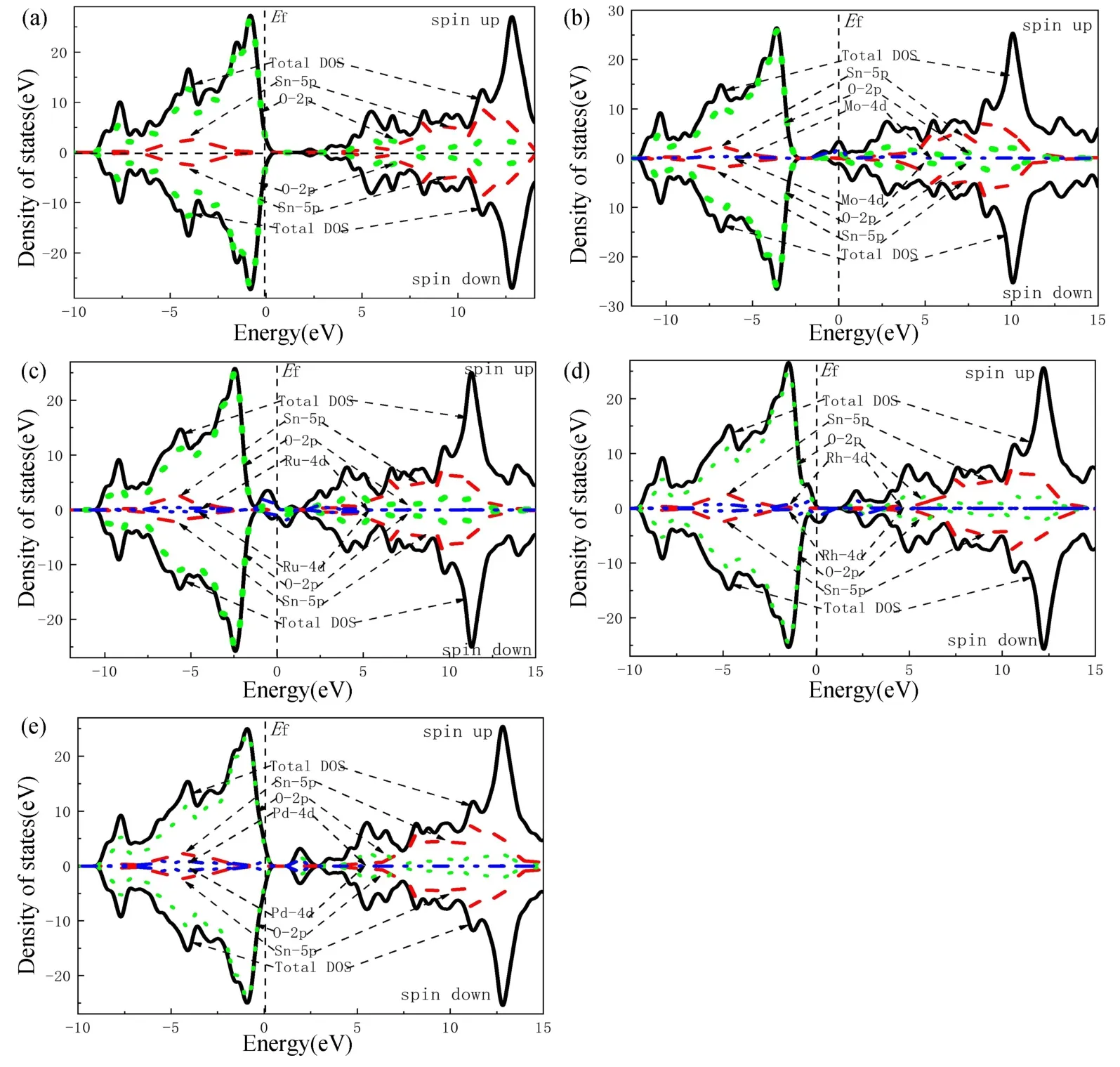
图3 能量密度图。(a)纯SnO2;(b~e)Sn0.9375X0.0625O2(X=Mo/Ru/Rh/Pd)Fig.3 Density of states.(a)Pure SnO2;(b-e)Sn0.9375X0.0625O2(X=Mo/Ru/Rh/Pd)
从图3(b)~(d)可知,Sn0.9375X0.0625O2(X=Mo/Ru/Rh)体系的总态密度均出现了自旋劈裂,导带与价带均由Sn-5p 态、O-2p 态和X(Mo/Ru/Rh)-4d 态杂化构成,并且各个体系自旋向上和向下电子密度的峰值都十分接近;在价带区(费米能级处)自旋向上和向下电子密度出现偏移,显然,这种偏移与X(Mo/Ru/Rh)-4d 态电子有关(与能带图分析一致),X(Mo/Ru/Rh)-4d 态与Sn-5p 态和O-2p 态相互发生自旋极化,也就说明体系将产生净磁矩而呈现铁磁性。而Mo-4d 态在导带区的底部上自旋态(费米能级处)提供了一个杂质峰,Ru-4d 和Rh-4d 态在下自旋态提供了一个杂质峰,使Mo 和Ru 的费米能级都穿过导带以及Rh 的费米能级穿过价带,同时拓宽了导带和价带的宽度,局域性增强,导电性增强[31];掺杂体系的电子轨道完全被X(Mo/Ru/Rh)-4d 态占据,出现了自旋劈裂,由于过渡金属4d 轨道的引入,使其与附近的原子产生强烈耦合作用而产生局域磁矩,从微观上验证了过渡金属掺杂(Mo/Ru/Rh)是引起SnO2体系产生磁性的主要原因;再根据自旋极化率定义[32],可知这三种体系自旋极化率接近100%。
如图3(e)所示,Sn0.9375X0.0625O2(X=Pd)体系的导带与价带均由Sn-5p 态、O-2p 态和Pd-4d 态杂化构成;价带区和导带区自旋向上和向下电子密度完全对称,未出现自旋劈裂,表明Sn0.9375X0.0625O2(X=Pd)体系无磁性,还可以看出费米能级略微穿过价带。
2.3 能带结构
为了更进一步分析纯SnO2和X(Mo/Ru/Rh/Pd)掺杂SnO2磁性的起源,研究了五者的自旋向上和向下的能带结构,并且它们都有其对应的能带宽度,G、F、Q、Z、G 为布里渊区高对称点,0 eV 为费米能级,当高于或低于费米能级很远时,能级变得非常平缓,属于深能级,不具备参考价值。因此,主要研究费米能级附近-10~10 eV 的能带结构。
从图4(a)可知,金红石相的SnO2在价带顶与导带底处自旋向上和向下的能带宽度都为Eg=1.318 eV,与Bakht 等[33]、Feng 等[34]和Li 等[35]的计算结果1.310 eV,1.230 eV 和0.980 eV 都非常相近,说明本实验数据是可信的。但它们都小于实验值3.600 eV[36],这是因为在半导体材料的本征和掺杂下能带宽度一般都要小于它所对应实验值的30%~50%,这主要是由于密度泛函理论里的LDA 引起的,低估了Sn的5s、5p 态与O 的2p 态之间的排斥作用,由于只考虑能量的相对变化,这种误差不会影响结果的可靠性。纯SnO2自旋向上和向下的能带完全吻合,未产生自旋劈裂,无磁性(与态密度分析一致);导带最低点和价带最高点均在布里渊区的G 点,说明纯SnO2为直接带隙半导体材料。
图4(b)为单掺Mo 的SnO2能带结构,自旋向上和向下的费米能级都进入导带,属于n 型导电(与态密度结果一致),且它们的带隙值分别为1.251 eV 和1.375 eV,呈现半金属性;在价带顶、价带的低能区以及导带的高能区自旋向上和向下的能带结构一致;而在导带底的能级出现能级劈裂,在-1.5~1.0 eV 之间,有3 条能级不对称,表明能带产生了自旋劈裂,从而产生磁性,说明磁性主要来源于Mo 原子。
图4(c)为单掺Ru 的SnO2能带结构,自旋向上和向下的能带值分别为1.506 eV 和1.532 eV;在自旋向上处,出现3 条杂质态能级,3 条位于价带顶,且自旋方向一致,在自旋向下处,出现1 条杂质态能级,1条位于价带顶,自旋方向一致,说明价带顶与导带底之间不再是空隙带,出现了一部分杂质能级,使电子更易跃迁,提高光催化效率。在价带顶、价带的低能区以及导带的高能区自旋向上和向下的能带结构一致;而导带底出现能级劈裂,在0~1 eV 之间,自旋向下的能级有2 条,与自旋向上能级不对称,表明能带产生自旋劈裂,从而产生磁性,与图3(c)结果一致,磁性主要来源于Ru 原子的掺入。
图4(d)为单掺Rh 的SnO2能带结构,自旋向上和向下的能带值分别为1.369 eV 和1.412 eV;在自旋向下处,出现2 条杂质态能级,1 条位于价带顶和1 条位于导带底,自旋方向一致,说明价带顶与导带底之间不再是空隙带,出现了一部分杂质能级,使电子更易跃迁。在导带高能区自旋向上和向下的能带结构一致,而导带底的能级出现能级劈裂,在0.5~2.5 eV 之间,有2 条能级不对称;在价带低能区自旋向上和向下的能带结构一致,而价带顶出现能级劈裂,在-0.5~0 eV 之间,自旋向上的能级有2 条,与自旋向下能级不对称,表明能带产生自旋劈裂,从而产生磁性,说明磁性主要来源于Rh 原子,与Bouamra 等[37]研究的结果一致。
图4(e)为单掺Pd 的SnO2能带结构,在价带顶与导带底处的自旋向上和向下的能带宽度都为Eg=1.512 eV,且自旋向上和向下的能带结构一致,能级对称,说明体系无磁性。
综合图4(b)~(e)可知,与纯SnO2相比,自旋向上和向下的能级集体往深能级方向移动,能级均变得密集,能级起伏均变小,电子轨道重叠程度均变大,说明局域性增强,导电性增强;而掺杂后的体系,都为直接带隙半导体材料。

图4 能带结构图。(a)纯SnO2;(b~e)Sn0.9375X0.0625O2(X=Mo/Ru/Rh/Pd)Fig.4 Band structure.(a)Pure SnO2;(b-e)Sn0.9375X0.0625O2(X=Mo/Ru/Rh/Pd)
2.4 自旋电子态密度
由图3 和图4 可知,纯SnO2和Sn0.9375Pd0.0625O2体系无磁性,所以没有自旋电子态密度图。图5 为Sn0.9375X0.0625O2(X=Mo/Ru/Rh)体系的自旋电子密度图,可以看出大多数自旋密度集中在掺杂剂X(Mo/Ru/Rh)原子和第一个近邻的O 原子中,远距离的O原子贡献很小(与表2 磁矩分布一致),也就是大多数磁矩限制在X(Mo/Ru/Rh)掺SnO2的八面体内,但X(Mo/Ru/Rh)与其周围六个O 原子之间的FM 耦合可能有助于将FM 相互作用扩展到更远的原子上,这是p-d 轨道耦合交换的直接证据,X(Mo/Ru/Rh)原子是磁矩的主要贡献者,O 原子发生轻微极化,X(Mo/Ru/Rh)原子发生自旋极化(与图3 结果一致)。

图5 自旋电子密度图。(a~c)Sn0.9375X0.0625O2(X=Mo/Ru/Rh)Fig.5 Spin electron density.(a-c)Sn0.9375X0.0625O2(X=Mo/Ru/Rh)
为了进一步确定掺杂体系的磁性状态,分别对自旋向上和向下电子密度进行定值计算。软件中2*integrated spin density/μB是体系的电子自旋净磁矩,2*integrated | spindensity| 是电子自旋密度的模[37-38]。当二者不为0 且相等时,电子自旋方向一致,体系为铁磁;当前者为0 后者不为0 时,相邻的电子自旋方向相反,体系为反铁磁;二者都为0 时,体系为顺磁(无磁)[28]。由计算结果可得,纯SnO2和Sn0.9375Pd0.0625O2体系引起的自旋磁矩为 0,表明纯 SnO2和Sn0.9375Pd0.0625O2体系是顺磁体。当X(Mo/Ru/Rh)掺入时,体系出现净磁矩分别为1.9530μB,1.9999μB,1.0000μB,但其数值都小于电子自旋密度的模(2.0561,2.1063,1.1393),表明掺杂后的SnO2是亚铁磁体,并且有良好的铁磁性质,因此X(Mo/Ru/Rh)掺杂SnO2体系是具有开发潜质的DMS 材料。
为了验证上一步分析,体系表征为铁磁(FM)或反铁磁性(AFM),还可以观察能量差ΔE=EAFM-EFM[39]。如表2 所示,通过计算得出体系的配置,纯SnO2只有本征能量,体系无磁,总磁矩为0。X(Mo/Ru/Rh)掺杂SnO2体系,ΔE分别为0.0032,0.0021 和0.0022 eV,表明体系为铁磁性,进一步分析掺杂后引起的自旋极化,可知体系主要的磁矩来自于X(Mo/Ru/Rh)原子以及周围被极化了的Sn 原子和O 原子。其中X(Mo/Ru/Rh)原子磁矩分别为1.62μB,1.38μB和0.59μB,总磁矩分别为2.06μB,2.11μB和2.11μB,在误差允许范围内为玻尔磁子的整数倍,这是铁磁半金属[40]的一个重要特性(与图4 结果一致)。可以发现,Sn 原子和O 原子对磁矩的贡献与其所处位置有很大关系,在X(Mo/Ru/Rh)原子周围的六个O 原子中,磁矩分别在-0.02μB~0.02μB,-0.01μB~0.01μB和-0.01μB~0μB之间,而两个Sn 原子中,磁矩分别在-0.02μB~0.05μB,0μB~0.10μB和0μB~0.12μB之间,说明X(Mo/Ru/Rh)和周围的Sn 原子和O 原子发生了相互交换耦合作用(与图5 结果一致)。因此X(Mo/Ru/Rh)、Sn 和O 原子的交换耦合作用是磁性的一个重要来源。由于Mo、Ru 和Rh 离子注入后,SnO2由无磁材料转变为磁性材料,这些结果表明,SnO2半导体可以作为实现自旋电子器件的母体材料。

表2 所有计算模型的磁特性Tab.2 Magnetic properties of all compute models
Pd 掺杂SnO2体系,ΔE为0.0013 eV,基于理论,体系为铁磁性,但进一步分析掺杂Pd 原子引起的自旋极化,发现Pd 原子磁矩为0μB,Sn 原子磁矩为0μB,O 原子磁矩为0μB,总磁矩为0.58×10-3μB,在误差允许范围内为零磁矩,所以只看ΔE数据还不完整,还要看体系磁矩和2*integrated spin density/μB的值,综合得出Pd 掺杂的体系是无磁性的。
由此可见,Mo、Ru 和Rh 掺杂SnO2体系可获得室温铁磁性,总自旋磁矩越来越小,根据泡利不相容原理,Mo-4d 态的前4 个电子和Ru-4d 态前6 个电子的自旋方向相反使它们诱导的磁矩相互抵消,还有1个电子诱导磁矩的产生;Rh-4d 态前8 个电子自旋方向相反使得它们诱导的磁矩相互抵消,但体系由于d轨道没有占满,存在一些磁矩;Pd-4d 态的10 个电子自旋方向相反使得它们诱导的磁矩相互抵消,导致其掺入SnO2后的净磁矩为零。因此过渡金属元素4d 轨道的单电子是诱导极化净磁矩的关键。
X(Mo/Ru/Rh/Pd)掺杂SnO2体系的居里温度TC可以用经典的Heisenberg 模型在平均场近似下使用布里渊区函数表达式[41]来估算得出,即:

式中:kB是玻尔兹曼常数;x是掺杂原子的浓度。根据表2 数据可知: 每种配置的TC都和室温相差不大,其中Mo 掺SnO2的居里温度最高,达到396 K,说明可得到高于室温的稀磁半导体材料,这将为设计和制备Mo 掺杂SnO2系统的磁学性能提供理论依据。
3 结论
本文基于密度泛函理论下的第一性原理平面波超软赝势方法,对过渡金属(Mo,Ru,Rh,Pd)掺杂SnO2的电子结构与磁学性质进行了计算与分析,得出了以下结论: Sn—O 的键布局数随着原子的电荷数的增加而呈现出增大的趋势,表明体系的空穴载流子在增加,共价性也在增强,磁交换作用变强。过渡金属(Mo/Ru/Rh)掺杂SnO2体系自旋向上和向下的能带和态密度图在费米能级处均不对称并且显示出半金属性质,说明体系有净磁矩产生,与Bouamra 等研究结果相似,并且自旋电子态密度图验证了这一观点;其中Mo-SnO2的居里温度高于室温且稳定,有望制成稀磁半导体材料应用于电子自旋器件中。此外,通过对磁矩的分析,阐明了体系的磁性主要是由掺杂原子的4d轨道所引入。
