抗病毒药物中间体替诺福韦的合成工艺研究
2023-01-09尚胜捷陈云峰
尚胜捷,王 璨,陈云峰
武汉工程大学化学与环境工程学院,湖北 武汉 430205
替诺福韦(tenofovir,PMPA)于1993年被发现具有抗核苷酸逆转录的作用,具有很好的抗病毒效果[1],但是由于其使用过程中具有肾毒性等副反应问题,基于此作为前药,陆续开发了一些新的核苷类抗病毒药物,吉利德公司开发的富马酸丙酚替诺福韦(tenofovir alafenamide fumarate,TAF)和富马酸替诺福韦酯(tenofovir disoproxil fumarate,TDF)分别于2016年和2008年被FDA批准用于治疗乙肝(hepatitis B virus,HBV)和人类免疫缺陷病(human immunodeficiency virus,HIV),其在体内通过代谢形成R-9-(2-磷酸甲氧基丙基)-腺 嘌 呤[R-9-(2-phosphonylmethoxypropyl)-adenine,PMPA]的前药而发挥抗病毒作用,值得注意的是,它们的化学合成的核心中间体为PMPA[2]。另外,近期也有报道PMPA可直接用于治疗严重急性呼吸征冠状病毒感染[3-5]。鉴于PMPA在医药领域的重要性,其合成工艺备受关注。
1999年,吉利德公司首次报道了合成PMPA的商业路线[6],总收率为13%,随后经过优化,使总收率提高到24%(图1)[7-8]。整条反应路线是以腺嘌呤和碳酸丙烯酯为起始原料的,然而由于腺嘌呤分子上的6号位氮原子和9号位氮原子竞争,反应的选择性较差,最终导致目标分子N9-(2-羟基丙基)腺嘌呤(5)产率降低。1990年,Proenc等[9-10]报道了以二氨基马来腈(1)为起始原料,合成咪唑衍生物的方法,在此基础上,2020年,Derstine等[11]报道了以1为起始原料合成5的非核苷路线,该条路线巧妙的避免了6号位氮原子和9号位氮原子的竞争,手型异丙醇骨架在合成咪唑环前被安装到分子中,并且保留了手型结构,这个方法为合成PMPA提供了新的选择。同时,该方法也有较大的优化空间,相关的产物后处理较为繁琐不适于工业生产,可以对后处理进行优化,使其更适合工业化生产;另外,作者使用较为昂贵的三甲基溴硅烷(trimethylbromosilane,TMSBr)等试剂,可以通过优化反应条件,使用更为经济的试剂,有利于降低合成成本。

图1 吉利德公司报道的替诺福韦合成路线Fig.1 PMPA synthesis route reported by Gilead
从5合成PMPA的方法也有较多报道。2010年,Brown等[12]使用叔丁醇镁作为碱,将5烷基化(图2式1);在第一步过程中,烷基化可以在极性非质子溶剂中进行,其中,N,N-二甲基甲酰胺(N,N-dimethylformamide,DMF)、N-甲基吡咯烷酮(N-methylpyrrolidone,DMP)效果最好,但由于DMF存在游离的二甲胺,会导致一些二甲酰磷酰胺产生,故作者选用NMP作溶剂。在第二步合成过程中,作者使用路易斯酸催化亚磷酸二乙酯脱烷氧基,用三甲基氯硅烷(trimethylchlorosilane,TMSCl)代替较昂贵的TMSBr;在此步骤中,并不需要分离出中间产物N9-[(2-羟甲基亚磷酸二乙酯)丙基]-腺嘌呤(8)。2016年,Riley等[13]使用甲基格式试剂和叔丁醇体系,将5烷基化(图2式2);在第一步烷基化过程中,由于叔丁醇镁的价格过高,并且过量镁离子与8络合形成少量副产物,因此作者使用甲基格氏试剂代替叔丁醇镁作为碱。作者并没有对亚磷酸试剂进行筛选,是因为Mg2+与8络合,使得5更容易烷基化[14]。使用环己烷做为溶剂,更容易提纯中间产物8。在第二步水解过程中,作者使用氢溴酸和醋酸体系做水解。综上所述,因为氢溴酸的毒性和反应体系的除水难度,以及三甲基溴硅烷是一种较为昂贵的水解试剂,本文使用已经报道过的TMSCl和溴化钠(sodium bromide,NaBr)体系先把对甲苯磺酰基甲氧基亚膦酸二乙酯(9)水解[11],再进行5的烷基化,降低了反应危险和生产成本,适合工业生产。
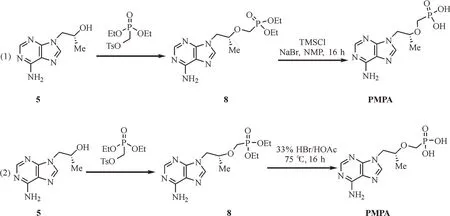
图2 替诺福韦的合成路线Fig.2 Synthesis route of PMPA
为了解决传统合成工业遇到的问题和提供可供选择的工艺合成路线,通过文献对比,本文选择1为起始原料,避免使用腺嘌呤来合成PMPA;另外,最后一步合成中使用更经济的碱和优化反应的后处理,进一步控制合成成本。综上所述,选择以下合成方案(图3)。

图3 替诺福韦的合成方案Fig.3 Proposed synthesis scheme of PMPA
1 实验部分
1.1 化学试剂及材料
1.1.1 主要化学仪器Varian 600 MHz以及400 MHz核磁共振仪(TMS做内标);RY-1型熔点仪;FINNIGAN TRACE MS和BRUKER APEX IV FTMD质谱仪;DF101S集热式恒温加热磁力搅拌器;IKA旋转蒸发仪。
1.1.2 化学试剂二氨基马来腈(纯度≥98%,国药试剂);异丙醇胺(纯度≥98%,优索试剂);氢氧化钡(纯度≥98%,天津大茂化工试剂);醋酸甲脒(纯度≥98%,麦克林试剂);三甲基氯硅烷(纯度≥99%,麦克林试剂);叔丁醇钠(纯度≥98%,国药试剂);对甲苯磺酸酰基甲氧基亚磷酸二乙酯(纯度≥98%,麦克林试剂);氯化镁(纯度≥98%,国药试剂);N,N-二甲基甲酰胺(纯度≥99%,天津富宇试剂);乙腈(纯度≥99%,天津富宇试剂)。
1.2 实验方法
1.2.1 N-亚甲胺基-(2-氨基-1,2-二氰基乙烯基)乙酯(2)的合成称取20 g原料1于250 mL的烧瓶中,加入80 mL的乙醇,常温下搅拌5 h。再将25 mL原甲酸三乙酯一次性滴加进反应中。将126.5 mg的三氟乙酸(trifluoroacetic acid,TFA)加入反应中(注意此过程需要严格控制TFA的量,若滴加过量,则会导致产物粘结而难以搅拌)。反应在油浴锅中加热至40℃反应2 h。反应完毕后,在20 min内冷却至室温,继续冷却至反应内部温度0℃。通过真空过滤分离固体,得到的固体在真空下干燥。反应最终得到26.2 g浅灰色固体,产率为94.2%。1H NMR(400 MHz,DMSO-d6)δ8.21(s,1H),4.05(s,2H),3.57(s,3H)。13C NMR(100 MHz,DMSO-d6)δ157.37,123.53,115.26,115.12,98.79,55.10。
1.2.2 N-(2-羟丙基)-4-氰基-5-氨基咪唑(4)的合成在250 mL的圆底烧瓶中加入14.4 mL异丙醇胺,用20 mL乙腈溶解,室温下搅拌5 min。将20 g原料2溶解于140 mL乙腈中,并一次性加入反应中,此时反应液变为深红色,用额外12 mL乙腈冲洗瓶壁上的残留固体反应2.5 h。原料2消耗完毕后,使用冰水浴将反应混合物冷却至0~5℃,加入30.4 g氢氧化钡和72 mL蒸馏水。随后将反应加热至40℃,反应液变为深棕色,反应1.5 h。反应完全后,反应混合物通过布氏漏斗过滤,在40℃下减压脱去溶剂,产物以黑油状液体留在烧瓶中。在黑油产物中添加氢氧化钡的饱和800 mL乙醇溶液,常温下搅拌过夜(约12 h)。抽滤反应液除去钡盐沉淀,40℃下减压脱去溶剂,得到的固体在真空下干燥,最终得到浅灰色固体产物18 g,产率81.4%。1H NMR(400 MHz,DMSO-d6)δ7.11(s,1H),6.08(s,2H),5.06(d,J=4.9 Hz,1H),3.87~3.74(m,2H),3.68~3.58(m,1H),1.05(d,J=6.2 Hz,3H)。13C NMR(100 MHz,DMSO-d6)δ148.37,134.00,118.07,90.71,65.36,50.62,21.07。
1.2.3 N9-(2-羟基丙基)腺嘌呤(5)的合成在250 mL圆底烧瓶中加入27.8 g原料4和醋酸甲脒29.5 g,将混合物溶于100 mL的DMF中。加热混合物至内温100℃,反应过夜(约12 h)。反应完全后,关闭加热并加入异丙醇150 mL,将反应液冷却至室温(25℃)。再将反应液冷却至-15℃的内温,在此温度下搅拌3 h。过滤悬浮液,用80 mL冷的异丙醇洗涤固体,将分离出的固体置于真空烘箱中干燥2 h,最终得26.5 g白色固体产物5,产率82.0%,熔点228~235℃(文献值:230℃)。1H NMR(400 MHz,DMSO-d6)δ8.02(s,1H),7.99(s,1H),5.03(d,J=4.3 Hz,1H),4.09(d,J=9.3 Hz,1H),4.04~3.94(m,2H),1.06(d,J=5.6 Hz,3H)。13C NMR(100 MHz,DMSO-d6)δ157.17,149.03,145.75,141.39,124.15,65.20,50.86,21.26.ESI-MSm/z:194.3。
1.2.4 对甲苯磺酰基甲氧基亚磷酸(6)的合成称取50 g对甲苯磺酰基甲氧基亚磷酸二乙酯溶解于78 mL的乙腈中,加入40 g NaBr和42 g TMSCl,将混合物在60℃的油浴中加热3 h。原料消耗完毕后,过滤钠盐,减压脱去溶剂,残留物用50 mL蒸馏水洗涤,加水过程中可以观察到游离的白色固体,混合物常温下搅拌过夜(约12 h)。得到的反应液用正己烷洗涤,保留水相。得到的水相中加入50 mL的甲苯共沸干燥,最终得到无色或淡黄色的油状产物39.0 g,其产率为94.4%。1H NMR(600 MHz,DMSO-d6)δ7.37(m,J=16.5,7.8 Hz,2H),6.97~6.86(m,2H),3.28(m,J=53.8,14.4,8.3 Hz,3H),2.01(d,J=23.5 Hz,3H)。13C NMR(100 MHz,DMSO-d6)δ145.51,131.59,130.06,128.19,64.05(d,J=6.5 Hz),21.61。
1.2.5 替诺福韦(PMPA)的合成向500 mL三颈烧瓶中加入400 mL的DMF,并通过冰浴使溶液降温至0℃,在此温度下加入60 g叔丁醇钠和6 g氯化镁,此时溶液中有部分叔丁醇钠未完全溶解。待溶液内温降至0℃时,一次性加入20 g原料5,搅拌10 min,期间反应液温度保持在5℃以下。在1 h内分4次加入6(每次添加12 g),注意保持反应液温度不超过5℃,且不宜过快添加,避免溶液变得黏稠而搅拌困难。添加完毕后将反应液升温至室温(约20℃),过夜反应(约12 h)。原料被大部分消耗完毕后,停止反应,减压脱去溶剂,剩余固体用浓盐酸调节pH至2左右,期间有白色固体析出,使用冰浴将反应液降温至5℃以下。过滤白色固体,再用200 mL正己烷洗涤固体,得到的白色固体置于75℃下真空干燥。最终得到白色固体19.3 g,产率65.0%,熔点180~188℃(文献值:180℃)。1H NMR(400 MHz,DMSO-d6)δ8.21(s,2H),7.13(s,2H),4.16~4.08(m,1H),4.02~3.98(m,1H),3.87~3.83(m,1H),3.81~3.69(m,2H),1.08(s,3H)。13C NMR(100 MHz,DMSO-d6)δ130.63,128.54,128.26,125.98,65.08,50.62,34.43,21.58,21.24.ESI-MSm/z:288.1。
2 结果与讨论
在合成2的过程中,使用原甲酸三甲酯或原甲酸三乙酯对最终产率影响不大,在两种原料市场价格相差不大的情况下,选择原甲酸三乙酯的原因在于反应副产物是乙醇,在绿色化学的要求下,相比于甲醇,生产更为安全,也容易处理。
从2到4的合成过程中,中间产物(3)不需要经过额外的分离,在不添加额外试剂的情况下,中间产物会缓慢成环生成4。加入额外的碱催化试剂(如碳酸氢钠、氢氧化钠、氢氧化钡等)会使得成环速率加快。日本Soda公司[15]使用氢氧化钠作为碱催化试剂,但是最终得到的产品聚合成黑油状,难以分离并且不能直接用于合成下一步。1990年,Proenc等[9]报道使用氢氧化钡作为碱催化剂,游离的钡离子与产物络合解聚,最终产品经过减压脱去溶剂呈浅灰色固体,后处理方式简单且方便继续做衍生化。采取此种方式,最终得到预期产品。
在合成5的过程中,溶剂应该选择极性非质子高沸点溶剂,水的存在会导致反应不佳,最终分离收率会降低,反应温度在80~120℃之间,过低的温度也会导致反应不佳,当反应内温高于120℃时,未能观察到产物的转化(结果如表1所示)。在后处理过程中,筛选了一些醇类溶剂,异丙醇表现出最好的分离效果。

表1 反应温度对分离收率的影响Tab.1 Effect of reaction temperature on isolation yield
在通过水解9合成6的过程中,通常使用路易斯酸作为水解试剂。Derstine等[11]报道使用TMSBr作为路易斯酸。2015年,Pizova等[16]报道了一种水解亚磷酸二乙酯的方法,作者使用TMSCl/NaBr体系水解。综合考虑原料的价格,选择TMSCl/NaBr水解体系,使得合成成本进一步降低。值得注意的是,6的纯度过低会影响5到PMPA的转化。
在合成PMPA的过程中,首先筛选了溶剂,溶剂的选择出于两方面考虑:一方面体系中的质子会消耗碱;另一方面,原料极性较大,小极性溶剂不能使原料有效溶解,从而导致5转化降低。因此考虑使用极性非质子溶剂,在此过程中尝试筛选1,4-二氧六环、乙腈、正己烷、DMF、NMP,其中DMF和NMP表现出更好的转换效果(结果如表2所示)。碱的选择是控制合成成本的关键因素之一,传统工业生产使用较为昂贵的叔丁醇镁,因此尝试使用叔丁醇钠,筛选了叔丁醇钠的物质的量作为对反应的转化影响因素,结果显示,5-7倍物质的量的叔丁醇钠有最好的转化效率,考虑到镁离子与6络合,会促进反应的转化,因此采用叔丁醇钠和氯化镁共催化的方法,结果表明更有利于PMPA的转化。温度的控制也是保证转化率的重要因素之一,在向反应液中添加原料6的过程中,若温度高于10℃,会导致反应液凝结而难以搅拌。另外,过快的滴加原料6,也会导致反应液凝结,最好的添加原料6的方式是在保持低温的前提下,缓慢滴加原料6的DMF溶液。

表2 反应溶剂对分离收率的影响Tab.2 Effect of reaction solvent on isolation yield
3 结论
PMPA的传统合成路线遇到的问题为:(1)以腺嘌呤为起始原料合成PMPA,在生成5过程中,不可避免的产生腺嘌呤上的N6-取代的烷基化副产物7。(2)从5到PMPA过程中,叔丁醇镁及三甲基溴硅烷等试剂的使用增加了合成路线的成本。考虑到上述问题,提出以下解决思路:(1)设计一种非核苷合成路线,避免使用腺嘌呤作为合成PMPA的起始原料。(2)考虑使用更廉价的碱、路易斯酸、溶剂等原料,从而控制合成成本。在已有的合成方法的基础上,本工作解决了传统工艺路线问题,合成PMPA的总收率从24%提升到37%;简化了后处理方式,同时使用更为经济的原料(如用叔丁醇钠替代叔丁醇镁、用TMSCl替代TMSBr等),进一步控制合成成本,适合工业生产。
