氧化铁纳米催化剂的生物合成及光催化性能研究
2022-12-19周宛欣
薛 珊,杨 涛,周宛欣,唐 璇
(西安石油大学 化学化工学院, 西安 710065)
0 引 言
纳米生物合成技术近年来得到了越来越多的关注,生物合成清洁、无毒、反应条件温和可控,是一种环境友好的方法[1-2]。此外,生物合成也对纳米粒子表面进行了生物修饰,具有高生物相容性、高分散性和高稳定性[3-4]。虽然近年来生物合成纳米颗粒取得了较大进展,但是所合成的纳米颗粒种类仍然较少,且多为简单的单质金属,并受到微生物种类、pH值、金属离子浓度、温度等因素的影响[5],限制了其产业化及实际应用。
Srivastava等[6]首次用铜绿假单胞菌细胞外单步合成银、铁和铑等纳米颗粒; 细胞内合成钴和锂纳米颗粒,且在整个研究过程中无需添加纳米颗粒稳定的表面活性剂。吕清[7]采用希瓦氏菌Shewanella loihica PV-4 生物合成铜纳米颗粒,研究表明低浓度的铜离子及高浓度的希瓦氏菌有利于合成,并且希瓦氏菌在胞内和胞外均能生成Cu NPs。杨阔[8]研究发现地衣芽孢杆菌HLS胞内合成粒径在10~20 nm 的Cu NPs,并推测硝酸盐还原酶可能影响菌株对Cu NPs的合成。 Abdel-Aziz等[9]证实了里诺琴氏伯克霍尔德菌(Burkholderia rinojensis)细胞中存在的有机分子在合成氧化镁纳米颗粒(MgO NPs)中作为封盖剂,也有助于阻止纳米颗粒的团聚。大量研究表明生物分子有助于化合物作为封盖剂和稳定剂附着在纳米颗粒上,提高纳米颗粒的稳定性和生物相容性,防止颗粒的团聚、电离和聚集,控制其大小和形状[10]。
Fe2O3禁带宽度窄,在氧化还原反应中活性高[11],因此常被用来催化降解有机污染物,特别是Fe2O3NPs,相比于微米级颗粒,由于其具有丰富的多孔结构,在催化氧化过程中具有更好的催化活性。本研究采用铜绿假单胞菌和贝莱斯芽孢杆菌合成Fe2O3NPs,采用铜绿假单胞菌和贝莱斯芽孢杆菌生物合成Fe2O3NPs,探究合成参数对合成样品的影响,对Fe2O3NPs进行表征分析,并探究其对甲基紫和异噻唑啉酮有机废水光催化降解性能。
1 实 验
1.1 试剂与仪器
药品与试剂:琼脂粉,蛋白胨,牛肉浸膏,氯化钠,无水三氯化铁,无水乙醇,甲基紫,异噻唑啉酮,盐酸羟胺,1,10-菲啰啉(一水合物)均为分析纯;实验用水为去离子水。
仪器:FT-IR(Nicolet Nuxes 670),SEM(Thermo scientific Apreo 2C),TEM(FEI Talos F200X)TGA(TG-SDTA 851),XRD(D8 ADVANCE),氮气吸附/脱附仪(ASAP202),紫外可见分光光度计(UV754N),紫外可见近红外分光光度计UV-Vis(Lambda950)。
铜绿假单胞菌和贝莱斯芽孢杆菌均由学校微生物实验室赠送。
1.2 细菌的培养及催化剂的制备
铜绿假单胞菌和贝莱斯芽孢杆菌在牛肉膏蛋白胨液体培养基,分别培养36 和48 h(30 ℃,140 r/min)。离心(6 000 r,5 min)收集菌体,无菌水洗涤两次,细胞沉淀用无菌水重悬,在不同的FeCl3浓度(0.5~ 2.5 mmol/L),时间(2~10 h),温度(25~ 45 ℃)和pH值(5.0~7.0)条件下反应。离心得到前驱体(由铜绿假单胞菌制备的前驱体记为前驱体@PA,贝莱斯芽孢杆菌制备的前驱体记为前驱体@BV),冷冻干燥后放入马弗炉中焙烧2h,对产品进行表征分析。由铜绿假单胞菌制备的催化剂记为Fe2O3@PA NPs,贝莱斯芽孢杆菌制备的催化剂记为Fe2O3@BV NPs。
1.3 结构表征测试
采用TAG2型热重分析仪,测定前驱体的热稳定性;采用NICOLET-5700型红外光谱仪,用KBr薄片法进行检测;采用D8 ADVANCE型X射线衍射仪测定材料的晶体结构;采用场发射扫描电镜和透射电子显微镜对材料的形貌进行分析;采用氮气物理吸附测试材料的比表面积,孔径;采用Lambda950型紫外可见光谱仪,测试催化剂的紫外可见光吸收曲线;采用X射线光电子能谱仪对纳米催化剂进行化学组成,以及元素的化学价态分析。
1.4 光催化性能测试
1.4.1 光催化降解甲基紫
催化剂0.03 g,均匀分散在50 ml的10 mg/L的甲基紫溶液中,暗反应搅拌30 min,打开蓝光光源,每30 min取样并过滤。使用紫外-可见分光光度计测定甲基紫溶液在590 nm的吸光度。根据公式(1)计算降解率:
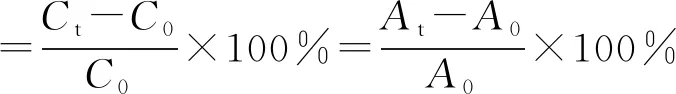
(1)
式中:C0是初始时刻甲基紫的浓度;Ct是t时刻甲基紫的浓度;A0为初始时刻甲基紫在最大吸收波长处的吸光度值;At为t时刻甲基紫在最大吸收波长处的吸光度值。
1.4.2 光催化降解异噻唑啉酮
取浓度为30 mg/L的异噻唑啉酮溶液(华科CMI∶MI=3∶1)50 mL,然后加入0.03 g的催化剂,通过暗反应搅拌30 min,打开紫外灯,每30 min取样并过滤。使用紫外-可见分光光度计测定异噻唑啉酮在273 nm的吸光度。根据公式(2)计算降解率:
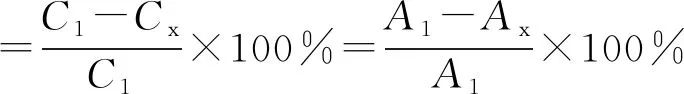
(2)
式中:C1是初始时刻异噻唑啉酮的浓度;Cx是x时刻异噻唑啉酮的浓度;A1为初始时刻异噻唑啉酮在最大吸收波长处的吸光度值;Ax为x时刻异噻唑啉酮在最大吸收波长处的吸光度值。
2 结果分析
2.1 制备前驱体的影响因素研究
2.1.1 Fe3+浓度与时间的影响
如图1,随着Fe3+浓度从0.5 mmol/L增大至2.5 mmol/L,该反应对铁离子的生物产率逐渐降低,在反应进行8 h后对Fe3+的产率几乎稳定。对铜绿假单胞菌而言,初始浓度为0.5和1.0 mmol/L的时候,8 h后产率达到77.50%以上,而初始浓度为2.5 mmol/L时,10 h时Fe3+的产率仅为54.44%;对贝莱斯芽孢杆菌而言,初始浓度为0.5和1.0 mmol/L的时候,8 h后产率达到80.20%,初始浓度为2.5 mmol/L时,10 h时Fe3+的产率为42.48%。考虑到0.5 mmol/L的Fe3+含量较少,因此选择Fe3+浓度1.0 mmol/L是最佳浓度,时间8 h的最佳反应时间。在高浓度的金属离子时,生物分子的数量可能不足以还原所有的金属离子,产率较低;金属离子浓度较低时,金属离子可以快速反应合成纳米颗粒,而最佳的反应时间与金属离子的浓度有关。

图1 Fe3+浓度与时间对产率的影响:(A) 铜绿假单胞菌; (B) 贝莱斯芽孢杆菌; 其中: (a)0.5 mmol/L; (b)1.0 mmol/L; (c)1.5 mmol/L; (d)2.0 mmol/L; (e)2.5 mmol/L
2.1.2 温度的影响
将反应温度控制在25~45 ℃,其对Fe2O3纳米光催化剂的影响如图2所示。
如图2所示,适宜的温度有利于提高反应速度[12],温度对产率的影响是先增加后降低,不同微生物的适宜温度也不同,温度低时微生物体内的蛋白质、酶等物质活性不高,产率较低;温度高时微生物体内的蛋白质酶变性,产率降低。铜绿假单胞菌在35 ℃时产率达到了79.86%,贝莱斯芽孢杆菌在40 ℃时产率为82.41%。铜绿假单胞菌和贝莱斯芽孢杆菌分别在35和40 ℃时合成前驱体的效果最佳。在合适的温度下,金属离子在成核过程中被快速消耗,纳米粒子的团聚减少,从而提高颗粒的形成和单分散。

图2 温度对产率的影响: (a) 铜绿假单胞菌; (b) 贝莱斯芽孢杆菌
2.1.3 pH值的影响
将反应体系的pH值控制在5.0~7.0之间,其对Fe2O3纳米光催化剂的影响如图3所示。
不同pH对反应的影响如图3。当pH值为5时,两种菌对Fe3+的产率都较低。铜绿假单胞菌在pH值为6.5时合成效果最好,产率达到了81.12%;而贝莱斯芽孢杆菌在pH值为7.0时合成效果最好,产率达到了82.41%。pH值改变生物分子的形状,而生物分子充当纳米粒子的封端和稳定剂[12]。超过合适的pH范围,由于某些负责生物分子的失活或降解引起的减少,会使得生成缓慢或者颗粒聚集。

图3 pH值对产率的影响: (a) 铜绿假单胞菌; (b) 贝莱斯芽孢杆菌
综上所述,铜绿假单胞菌制备前驱体的最佳条件是Fe3+浓度为C(Fe3+)=1.0 mmol/L、T=35 ℃、pH =6.5、t=8 h;贝莱斯芽孢杆菌制备前驱体的最佳条件是C(Fe3+)=1.0 mmol/L、T=40 ℃、pH=7、t=8 h。在最佳工艺条件下制备催化剂前驱体用于后续研究。
2.2 前驱体的热重分析
前驱体的热重分析曲线如图4所示。

图4 前驱体的热重分析: (a) 前驱体@PA; (b) 前驱体@BV
由热重分析图4可知,前驱物的重量在0~250 ℃范围内,前驱物的自由水和结合水逐步蒸发。在250~400 ℃范围内细胞结构快速分解灰化,重量明显下降。当温度升高到400 ℃左右后,前驱体的重量几乎不再发生变化。前驱体@PA的失重率约为58%,前驱体@BV失重率约为60%。此时,产物的晶型由无定型逐渐转化为Fe2O3。因此选择450 ℃作为焙烧温度。
2.3 催化剂的X-射线衍射分析
氧化铁纳米光催化剂的组分分析如图5所示。由图5可知,在煅烧温度为200 ℃时,X射线衍射图谱上并无明显的衍射峰,说明该条件下生成的产物为无定形非晶态。在煅烧温度为450 ℃时,2θ=33.22°、35.69°、49.44°、54.13°等位置都存在很强的衍射峰,与PDF85-0599的(104)、(110)、(024)、(116)的衍射峰位置基本符合。衍射峰强度高,结晶性好。X射线衍射分析表明了铜绿假单胞菌和贝莱斯芽孢杆菌生物法合成了Fe2O3纳米颗粒。
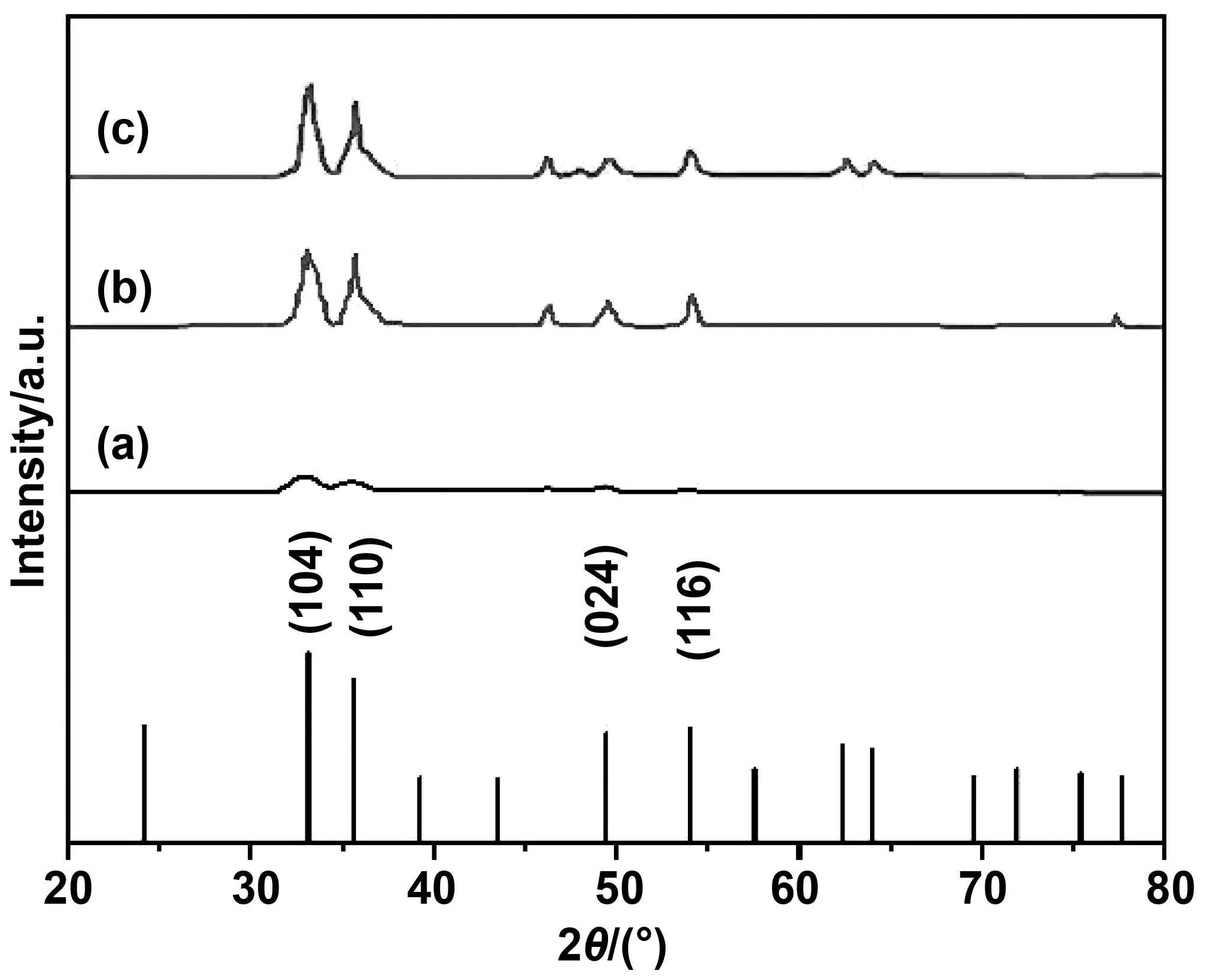
图5 Fe2O3的XRD图谱: (a) 200 ℃ 煅烧; (b) Fe2O3@PA; (c) Fe2O3@BV
2.4 催化剂的电镜分析
对不同的前驱体和催化剂进行SEM和TEM分析,如图6所示。前驱物@PA和前驱物@BV的SEM图6(a)(d)显示,合成的产品聚集在一起,产品中含有部分菌体,形状接近圆柱状,长度约1.5 μm。可以清晰的观察到纳米颗粒在细胞表面生成,表明这两种菌是胞外合成纳米颗粒。将前驱物焙烧使产品中的细胞灰化干净,得到纯纳米粒子,然后用扫描电子显微镜分析,结果如图6(b),(e)所示,可知纳米粒子为球形。焙烧后产物的TEM图6(c),(f)可以看到纳米粒子出现局部团结现象严重,这主要是与样品处理过程中的冷冻和焙烧退火有关。TEM图像显示Fe2O3@PA的平均粒径约13 nm,Fe2O3@BV的平均粒径约10 nm。

图6 不同前驱体和催化剂的电镜图: (a)前驱体@PA的SEM图;(b)Fe2O3@PA的SEM; (c)Fe2O3@PA的TEM; (d)前驱体@BV的SEM; (e)Fe2O3@BV的SEM; (f)Fe2O3@BV的TEM
2.5 催化剂的EDS分析
对催化剂进行EDS分析,结果如图7所示。从图得知Fe2O3@PA中的铁含量为38.25%,Fe2O3@BV的铁含量为45.29%,含有Si、P和S等元素是因为细胞的菌体成分。

图7 催化剂的EDS分析:(a) Fe2O3@PA; (b) Fe2O3@BV
2.6 催化剂的XPS分析
Fe2O3@PA和 Fe2O3@BV材料的全光谱表明材料存在Fe、O、Ca、C和P等元素。由图8 (B)中所示Fe2O3@PA的 Fe2p XPS 图谱可知Fe2p1/2 和 Fe2p3/2 的结合能分别为724.25和710.65eV,而图8 (D)中所示Fe2O3@BV的Fe 2p1/2和Fe 2p3/2的结合能分别为723.9和710.19eV,(B)和(D)在结合能为719.28和719.05 eV处还存在着一个特征峰,这是Fe2O3中Fe3+的特征峰[13,14]。进一步证明了生物法合成了Fe2O3纳米颗粒。
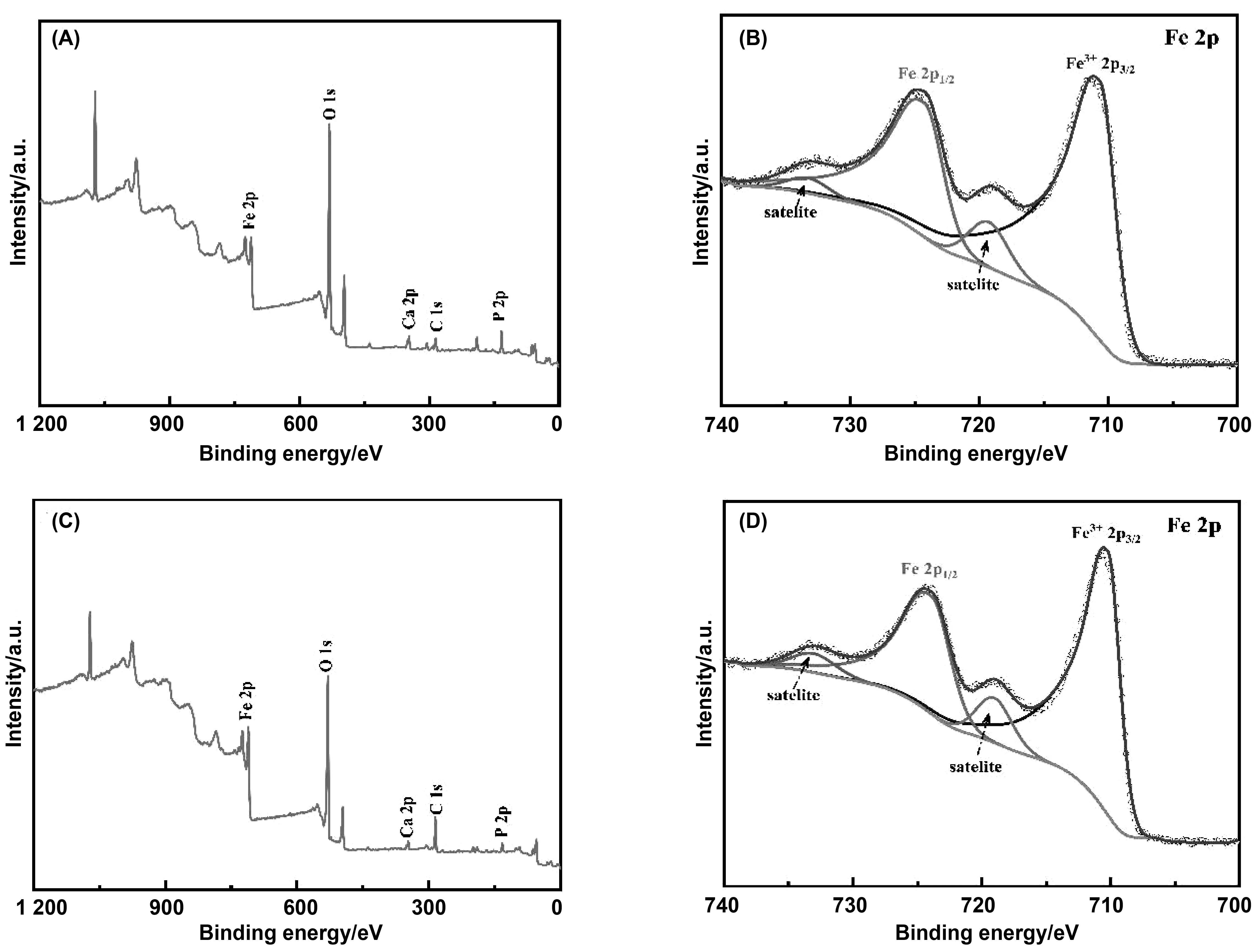
图8 Fe2O3的XPS图:(A) Fe2O3@PA全谱;(B) Fe2O3@PA Fe2p ;(C) Fe2O3@BV全谱;(D) Fe2O3@BV Fe2p
2.7 催化剂的傅里叶红外光谱分析
为了进一步研究氧化铁纳米光催化剂的结构,采用傅里叶红外光谱(FT-IR)对其进行表征。由图9可以得出,3 440 cm-1处是O-H的伸缩振动特征吸收峰,1 640 cm-1处是蛋白质亚甲基-C=O的振动吸收峰,1 382 cm-1处的谱峰,可能是酰胺C—N拉伸造成的。1 048 cm-1处为Fe-O键的伸缩振动吸收峰,620,547 与449 cm-1处属于Fe-O弯曲振动,是Fe2O3的特征吸收峰[15]。FT-IR光谱清楚地表明,细菌分泌的蛋白质等有机化合物参与了反应,也可能对细胞外产生的纳米颗粒起到稳定的作用。
2.8 催化剂的紫外漫反射分析
采用紫外可见吸收分光光度计对氧化铁纳米催化剂的紫外可见光吸收进行表征。由图10可知氧化铁纳米催化剂在紫外光区和小于500 nm的可见光区吸收性能好。 Fe2O3@PA纳米催化剂的吸收边在638 nm处,禁带宽度1.87 eV,Fe2O3@BV纳米催化剂的吸收边在655 nm处,禁带宽度1.85 eV,相较于商用的氧化铁[16]纳米催化剂(2.10 eV)禁带宽度减小。
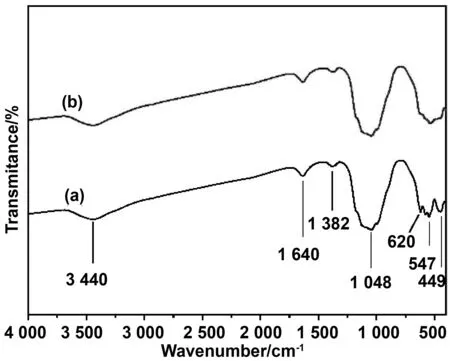
图9 Fe2O3的FT-IR图谱:(a) Fe2O3@PA; (b) Fe2O3@BV

图10 不同催化剂的紫外漫反射光谱:(A) 紫外漫反射光谱;(B)禁带宽度图; 其中: (a) Fe2O3@PA,(b) Fe2O3@BV
2.9 催化剂的氮气物理吸附
图11为不同样品的氮气吸附-脱附等温线以及孔径分布图。从图11中可以看到,催化剂的氮气吸附-脱附等温线,对应IV 型等温线[17],0.4~1.0 (P/P0)区间存在H3型滞后环,表明制备的材料中存在大量的狭缝介孔。由表1可知Fe2O3@PA的比表面积为24 m2·g-1,是商用Fe2O3的2.4倍,而Fe2O3@BV的比表面积为45 m2·g-1,是商用Fe2O3[16]的4.6倍。比表面积大,活性位点多,催化性能好。由孔径分布图可知Fe2O3@BV孔径较Fe2O3@PA的小,孔径分布大部分是属于0~2 nm的微孔结构,但是仍有一些数量的中孔和大孔结构。Fe2O3@PA含有大量的中孔结构,中孔和大孔的出现可能是由于高温过程中颗粒的团聚造成的。

图11 (A)不同样品的氮气等温吸附曲线; (B)不同样品的孔径分布曲线 其中: (a) Fe2O3@PA,(b) Fe2O3@BV
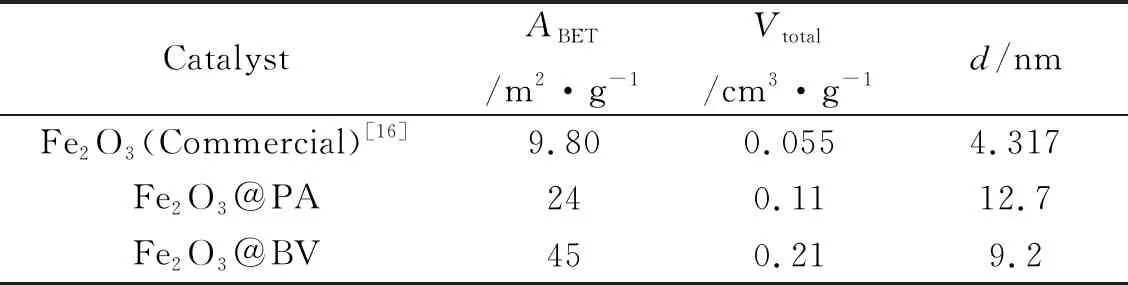
表1 Fe2O3的BET分析结果
3 光催化性能分析
3.1 光催化降解甲基紫
如图12可知,50 mL的 10 mg/L的反应液中不加催化剂作为空白对照,照射时间在5 h内,结晶紫溶液有降解,但是降解率仅12.75%;反应液中加入0.03 g催化剂,反应时间在0~3 h时是快速降解阶段,3 h以后降解速率降低,降解率曲线平缓上升,Fe2O3@PA在5 h内的降解率为61.29%,而Fe2O3@BV的最终降解率为69.87%。结果表明Fe2O3@BV的催化降解效果优于Fe2O3@PA。催化剂@BV的催化活性高的原因可能是:氮气物理吸附测试表明Fe2O3@BV的比表面积是Fe2O3@PA的1.9倍,比表面积大,活性位点多,催化活性高[18-19]。

图12 不同催化剂对结晶紫的降解率: (a)无催化剂; (b) Fe2O3@PA; (c)Fe2O3@BV; (d)Fe2O3(商用)
3.2 光催化降解异噻唑啉酮
图13是不同pH条件下样品对异噻唑啉酮的降解率。由图13可知只有紫外光照射时,异噻唑啉酮溶液也可以发生降解,研究认为异噻唑啉酮光转化主要有两种途径[20]:(1)主要是打开异噻唑啉酮环,经过一系列脱卤素、脱烷基和脱胺基化反应后最终裂解成更小的分子;(2)主要是异噻唑啉酮环受到光激发后,N-S 键断裂,形成自由基,进一步裂解成更小的分子。Fe2O3@PA和Fe2O3@BV半导体纳米催化剂的加入,光激发产生羟基自由基和空穴电子对,使异噻唑啉酮的降解率提高,紫外光和Fe2O3纳米催化剂的共同作用提高了异噻唑啉酮的降解速率,在酸性条件下降解速率较快,碱性条件下降解较慢。反应时间在0~3.5 h时是快速降解阶段,3.5 h以后溶液中异噻唑啉酮溶度减少,降解速率降低。Fe2O3@PA在pH值为5的条件下,4.5 h内降解率达到了76.21%, Fe2O3@BV的比表面积大,活性位点多,催化活性高,Fe2O3@BV的降解率达到了82.13%。

图13 不同pH条件下催化剂对异噻唑啉酮的降解率: (a)无催化剂,pH 5; (b)无催化剂,pH 7; (c)无催化剂,pH 9; (d) Fe2O3@PA, pH 5; (e) Fe2O3@PA, pH 7; (f) Fe2O3@PA, pH 9; (g) Fe2O3@BV, pH 5; (h) Fe2O3@BV, pH 7; (g) Fe2O3@BV, pH 9
4 结 论
采用两种不同的细菌,探究了Fe3+浓度、温度、时间及pH对合成产率的影响,制备出Fe2O3@PA和Fe2O3@BV NPs。FT-IR分析表明某些蛋白质分子可以阻止Fe2O3NPs的团聚,UV-Vis和BET分析表明生物法合成的Fe2O3NPs均优于商用。两种催化剂均对甲基紫和异噻唑啉酮溶液有明显的光催化活性,当光照射的Fe2O3半导体上,光激发产生电子-空穴对,迁移到半导体表面,氧化还原产生·OH自由基,有机物被氧化生成小分子的CO2和H2O等。在蓝光照射下,两种纳米颗粒对甲基紫的降解效果均优于常规方法制备的商用Fe2O3,5 h内Fe2O3@PA的降解率为61.29%,Fe2O3@BV的最终降解率为69.87%;紫外光和Fe2O3纳米催化剂的共同作用降解异噻唑啉酮,在pH=5的酸性环境下持续4.5 h,Fe2O3@PA的降解率为76.21%,Fe2O3@BV降解率能达到82.13%,pH=9的碱性环境下降解效果较差。
