密度泛函理论研究ZnGeP2 晶体中缺陷的稳定性及迁移机制*
2022-12-05马天慧雷作涛张晓萌付秋月布和巴特尔朱崇强杨春晖
马天慧 雷作涛 张晓萌 付秋月 布和巴特尔 朱崇强 杨春晖
1)(黑龙江工程学院材料与化学工程学院,哈尔滨 150050)
2)(哈尔滨工业大学化工与化学学院,哈尔滨 150001)
ZnGeP2 晶体是3—5 µm 中红外激光输出的最好频率转换材料,可实现激光器的全固态化和大功率输出.但在8—12 µm 处由于本证缺陷导致的吸收带与光参量振荡器的抽运波长交叠,限制了光参量振荡器的应用性能,使其无法实现远红外激光输出.本论文采用密度泛函理论讨论了ZnGeP2 晶体6 种缺陷结构的形成能与缺陷迁移机制.结果表明 VP和VGe 两种缺陷结构较难形成,四种缺陷容易形成.当Ge 原子微富余Zn 原子,温度为10 K,500 K和600 K时,当温度为273 K和400 K时,晶体的体积膨胀率与缺陷形成能的关系为负相关,即晶体体积膨胀率越大,缺陷形成能越低.差分电荷密度分析显示GeZn和VZn+GeZn 两种缺陷结构中原子间电子云密度增强,空位缺陷(VZn和VGe)与反位缺陷(GeZn和ZnGe)结合形成联合缺陷后,空位缺陷格点处电子云密度增强.当温度为10 K时,ZnGeP2 晶体的吸收光谱显示VGe,VZn,ZnGe和GeZn 四种缺陷结构在0.6—2.5 µm 有较明显吸收.VZn的迁移能最低,VGe 迁移能最高.VP 在迁移过程中迁移能与空间位阻有关,而VGe和VZn的迁移能与原子间距离有关.
1 引言
ZnGeP2(简称ZGP)晶体是目前通过频率转换产生中、远红外激光输出的最好的非线性光学材料之一,利用光参量振荡(OPO)、差频产生(DFG)以及光参量放大(OPA)等技术可实现中红外波段(3—5 µm)和远红外波段(8—10 µm)的可调谐连续激光输出[1,2].其在民用和国防领域均具有非常突出的应用:民用领域的应用包括红外激光诊疗、痕量气体监测、远距离化学传感、深空探测、工业生产过程检测和油田开采等;国防领域的应用包括红外激光干扰对抗、红外遥感、激光雷达、战场中生化武器甄别和作战目标仿真模拟等.但ZnGeP2晶体由于本征的点缺陷引起的光吸收和光散射,导致8—12 µm 处存在严重的双光子吸收,远红外激光输出效率较低[3−5].因此有必要对ZnGeP2晶体缺陷结构进行深入研究.
ZnGeP2晶体在合成和单晶生长中由于组分挥发,易形成空位点缺陷.ZnGeP2晶体中的缺陷类型可分为受主缺陷和施主缺陷两种类型,Rakowsky等[6]、Halliburton等[7]和Gehlhoff等[8]分别利用电子顺磁共振(EPR)和电子-核双共振验证了ZnGeP2晶体存在受主缺陷Zn 空位(VZn)、Ge 空位(VGe)和Zn 占Ge 位(ZnGe),受主缺陷是造成近红外吸收和能带边附近吸收的主要原因,其缺陷浓度1019—1020cm–3.实验表明ZnGeP2晶体含有的载流子浓度达到1010—1012cm–3,所以晶体中一定还存在大量的施主缺陷来补偿受主缺陷.Giles等[9]、Setzler等[10]和Gehlhoff等[11,12]分别利用EPR 数据证实了ZnGeP2晶体存在施主缺陷P 空位(VP)和阳离子反位缺陷GeZn.
Jiang等[13,14]采用基于第一性原理的全势能线性组合法(FP-LMTO),以费米能级为自变量,计算了ZnGeP2晶体中带有不同电荷的VP,VGe,VZn,ZnGe和GeZn缺陷的生成能,发现VGe和VP的生成能较高,VZn和GeZn缺陷的生成能较低,VZn与ZnGe复合缺陷不稳定.Jiang等[15]利用密度泛函理论和Hartree-Fock 方法研究了VZn在Jahn-Teller畸变中的自相互作用.
目前,ZnGeP2晶体的主要生长方法有垂直布里奇曼法和水平梯度冷凝法,缺陷结构的研究方法有电子顺磁共振、电子-核双共振、光学吸收、光致发光和霍尔效应等,消除或降低缺陷的方法有热退火、高能电子束或γ射线照射.ZnGeP2晶体的缺陷结构是一个庞大且复杂的体系,由于晶体的生长方法、生长条件以及研究缺陷条件的不同,都会造成研究结果的差异.随着理论计算的不断发展和完善,通过计算机模拟研究ZnGeP2晶体的缺陷结构不但可以验证实验结果,而且对实验研究提供可靠的理论指导.本论文采用基于密度泛函理论的第一性原理方法研究了ZnGeP2晶体六种缺陷结构(VP,VGe,VZn,ZnGe,GeZn+VZn和GeZn)的形成能及其与温度的关系.分析了缺陷超晶胞的差分电荷密度分布、缺陷的迁移机制及不同缺陷结构对晶体光学性能的影响.这对于建立晶体缺陷结构与性能的关系模型和探索降低缺陷密度的方法具有重要的意义.
2 计算模型与方法
ZnGeP2晶体为四方晶系,黄铜矿结构,空间群,是由2 个简单立方闪锌矿结构派生出来的,GeP4为正四面体结构,ZnP4为稍微变形的四面体结构.在电子结构和光学性能模拟计算中,交换-关联能选择广义梯度近似(GGA)中的Perdew-Burke-Ernzerhof(PBE)方法[16],静电势为超软赝势(USP)[17,18].动力学计算选择NPT 系综,即恒定压强和温度,压强为1 atm(1 atm=101.325 kPa),温度分别设定10 K,273 K,400 K,500 K和600 K.为了比较缺陷形成能大小,采用2× 1× 1 超胞计算形成能,缺陷密度为1/8,平面波截断能310 eV,k点数3×3×1.为更准确比较完美晶格和缺陷晶格原子间的作用力差别,采用2×1×1 超胞计算差分电荷密度分布.超晶胞光学性能计算导带数为60.
通过对空位缺陷过渡态的搜索,获得了ZnGeP2晶体中VP,VGe和VZn三种空位缺陷向邻近格点迁移过程中的迁移能,并找到迁移过程中的过渡态.VP迁移能计算采用2× 1× 1 超胞体系,VGe和VZn迁移能计算采用1× 2× 2 超胞体系.空位迁移的过程可以看作是对应原子迁移的一个反过程,在晶体中空位的邻近原子向空位迁移时必须要克服能量势垒才能完成此过程,这个必须克服的势垒即为空位迁移能,其计算公式为∆E=E2–E1,式中,E1为孤立空位的形成能,E2为过渡态能量,∆E为空位迁移能.
3 结果与讨论
3.1 ZnGeP2 晶体缺陷形成能

式中,∆Hf(Xq)为含有缺陷的超晶胞形成能,E(Xq)为含有缺陷的超晶胞总能量,E(per)为无缺陷超晶胞的总能量,q为超晶胞所带的电荷,µi为缺陷原子i的原子化学势,ni为超晶胞中增加或去除i原子的个数(增加为正,去除为负),EF为缺陷超晶胞的费米能级,EV为无缺陷晶胞价带顶能量,∆V为缺陷超晶胞与无缺陷超晶胞平均静电势之差,N为超晶胞中原子总数.

3.2 ZnGeP2 缺陷晶胞的体积变化率
图2 给出了10—600 K的2×1×1 超胞体积变化率V(T)/V0(T),V(T)为T温度时含缺陷晶胞体积,V0为T温度时无缺陷晶胞体积.图2显示晶体体积与温度之间没有严格的规律性,晶体体积变化率V(T)/V0(T)与温度无关,但温度升高,晶体体积有膨胀的趋势.图1和图2 对比发现,除VGe,其他5 种缺陷晶体体积的膨胀率与缺陷形成能负相关,即形成能越高膨胀率越低,VP,VZn(10 K,273 K,400 K),GeZn+VZn(10 K,400 K),ZnGe(10 K)和GeZn(10 K)缺陷晶体体积收缩,缺陷形成能均较高.

图1 缺陷形成能与温度的关系曲线Fig.1.Dependent curves of defect formation energy and temperature.

图2 ZnGeP2 缺陷晶胞体积变化率Fig.2.Volume change rates of defective cells for ZnGeP2.
图3为273 K和500 K 时ZnGeP2无缺陷晶胞和VP晶胞图,由图3 可知,当产生P 空位时,大部分VP邻近的P—Ge 键和P—Zn 键键长增大,导致晶格扭曲,但是含VP晶胞体积较完美晶胞体积略有减小(见图2).

图3 ZnGeP2 晶胞(a)273 K 完美晶胞;(b)273 K 时VP 晶胞;(c)500 K 完美晶胞;(d)500 K 时VP 晶胞Fig.3.Unit cells of ZnGeP2(a)Perfect cell at 273 K;(b)cell containing VP at 273 K;(c)perfect cell at 500 K;(d)cell containing VP at 500 K.
表1 所示为273 K和600 K 时完美晶胞与含阳离子反位缺陷晶胞中替换元素电荷和对应的键长.可以看出无论是Ge 替换Zn,还是Zn 替换Ge,替换后对应的Zn—P 键和Ge—P 键均比原来的Ge—P 键和Zn—P 键键长增大,导致晶格变形,含ZnGe和GeZn的晶胞体积较完美晶胞体积明显膨胀,替换后Zn的电荷显著增大,Ge的电荷明显降低,如273 K时,Zn的电荷由0.01 增大到0.13,Ge的电荷由0.69 降低为0.37,但替换前后P的电荷无明显变化.
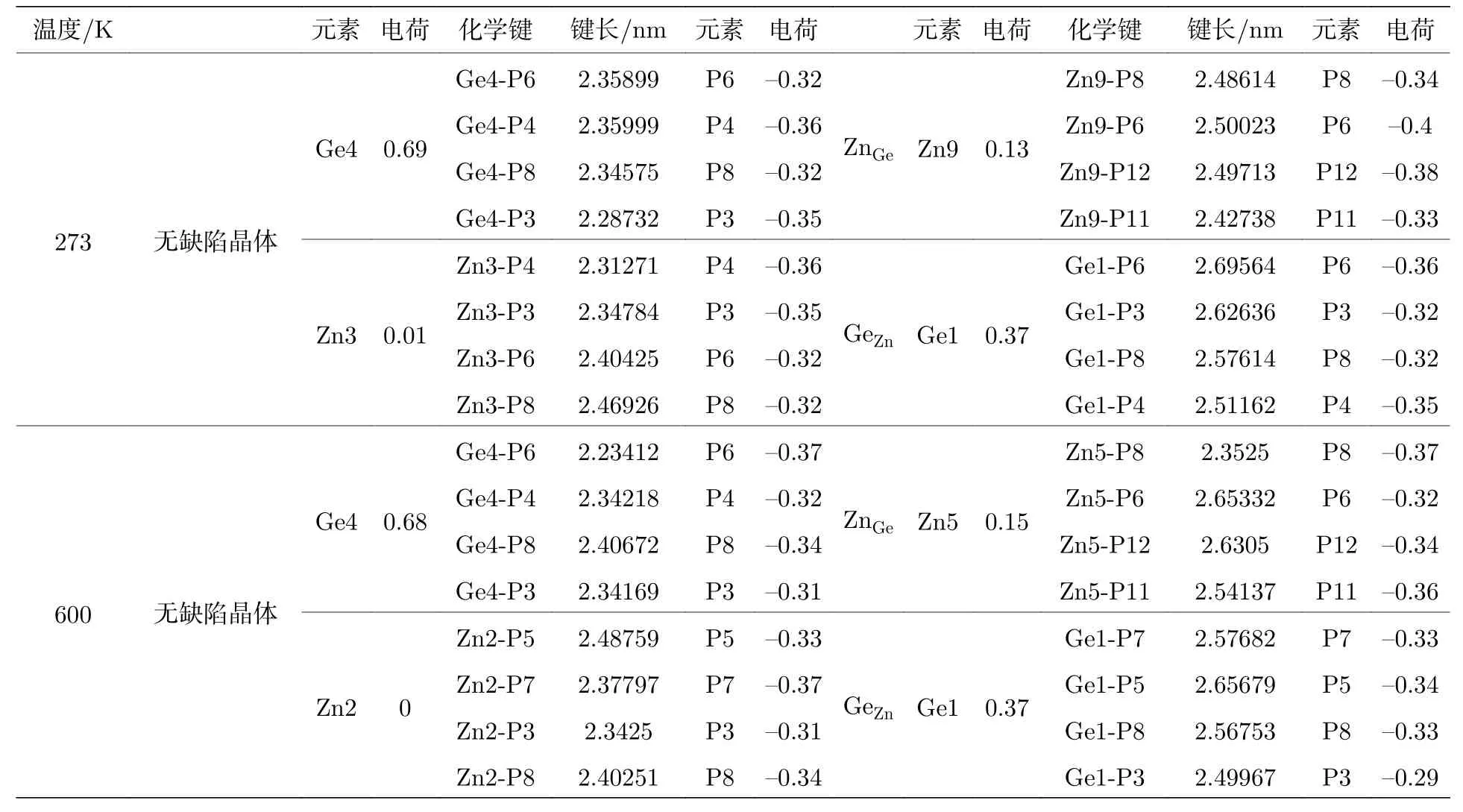
表1 含 Z nGe和G eZn的缺陷晶胞替换元素电荷和对应的键长Table 1.Charge of substitution element and corresponding bond length of defective cells containing Z nGe and G eZn.
3.3 ZnGeP2 缺陷晶胞的差分电荷密度分析
图4为ZnGeP2完美晶胞和缺陷晶胞在(200)晶面上的差分电荷密度分布图.与完美晶胞相对比,GeZn和VZn+GeZn两种缺陷结构导致Ge-Ge和Ge-Zn 原子间电子云密度显著增大,表明Ge替Zn 位使得其与周围原子作用力增强.孤立的空位缺陷VZn和VGe,缺陷格点位置电子云密度降低.但当空位缺陷与反位缺陷形成联合缺陷后,空位缺陷格点处电子云密度增强.比较VZn,VZn+GeZn和VZn+ZnGe三种缺陷结构,在VZn+GeZn联合缺陷中Zn 空位格点处电子云密度显著增强,同样VGe,VGe+GeZn和VGe+ZnGe相比较,VGe+GeZn联合缺陷中Ge 空位格点处电子云密度显著增强.
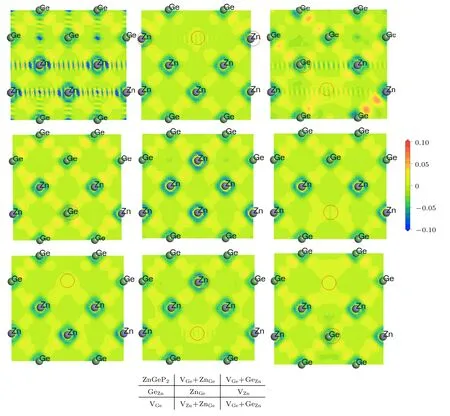
图4 无缺陷晶胞和含缺陷晶胞(200)晶面的差分电荷密度分布图(红色圆圈为缺陷位置)Fig.4.Differential charge density distribution of perfect cells and defective cells for(200)plane(red circles are defect positions).
3.4 ZnGeP2 缺陷晶胞的光吸收
图5为10 K 时ZnGeP2晶体6 种缺陷的吸收谱,其中VGe,VZn,Z nGe和GeZn缺陷在0.6—2.5 µm有较明显吸收,而VP和GeZn+VZn缺陷吸收较少,这与实验测试结果相符合,Setzler等[21]和Giles等[22]分别采用EPR 证实ZnGeP2晶体在1 µm和2.2 µm 处的光学吸收是由受主缺陷VZn造成的.

图5 ZnGeP2 缺陷晶胞的吸收谱Fig.5.Absorption spectra of defective cells for ZnGeP2.
3.5 ZnGeP2 晶体中的缺陷迁移机制分析
3.5.1 P 空位迁移
图6 给出(010)面P 原子的位置,并标记出P1 原子周围P2—P9 原子的位置.表2 给出P1 原子空位向P2—P9 迁移的迁移能和原子间距.

图6 (010)面P 原子标记图(采用2× 1× 1 超胞体系)Fig.6.Map of positions of P atoms for(010)plane(super cells of 2× 1× 1 are used).
表2 数据显示迁移能与原子间距离无明显正比例关系,P1-P3 原子间距离最大,但迁移能并不是最大,迁移能最大的是P1-P4 原子;P1-P8 迁移能最小,原子间距离并不是最小,原子在迁移过程中空间位阻可能是重要因素.
计算了P1 原子空位分别向P2,P3,P4,P5,P6,P7,P8和P9 迁移的正向和反向迁移能,每一组正向、反向迁移能并不相同.根据表2 中数据,P1 原子空位可以向P3,P6和P9 原子位置迁移,其正向迁移能小于逆向迁移能,迁移难度顺序为P1-P9 >P1-P3 >P1-P6.剩下5 种迁移方式倾向于逆向迁移,迁移难度排序为:P4-P1 >P5-P1 >P2-P1 >P7-P1 >P8-P1.比较迁移能数据发现P1-P8 正向和逆向迁移,迁移能都是最小的,因此在ZnGeP2晶体(010)面P 空位的迁移方向为P1 空位向P8 迁移或P8 空位向P1 迁移.通过计算找到8 种迁移方式的过渡态,图7 给出P1-P4,P1-P6,P1-P8 迁移的过渡态.
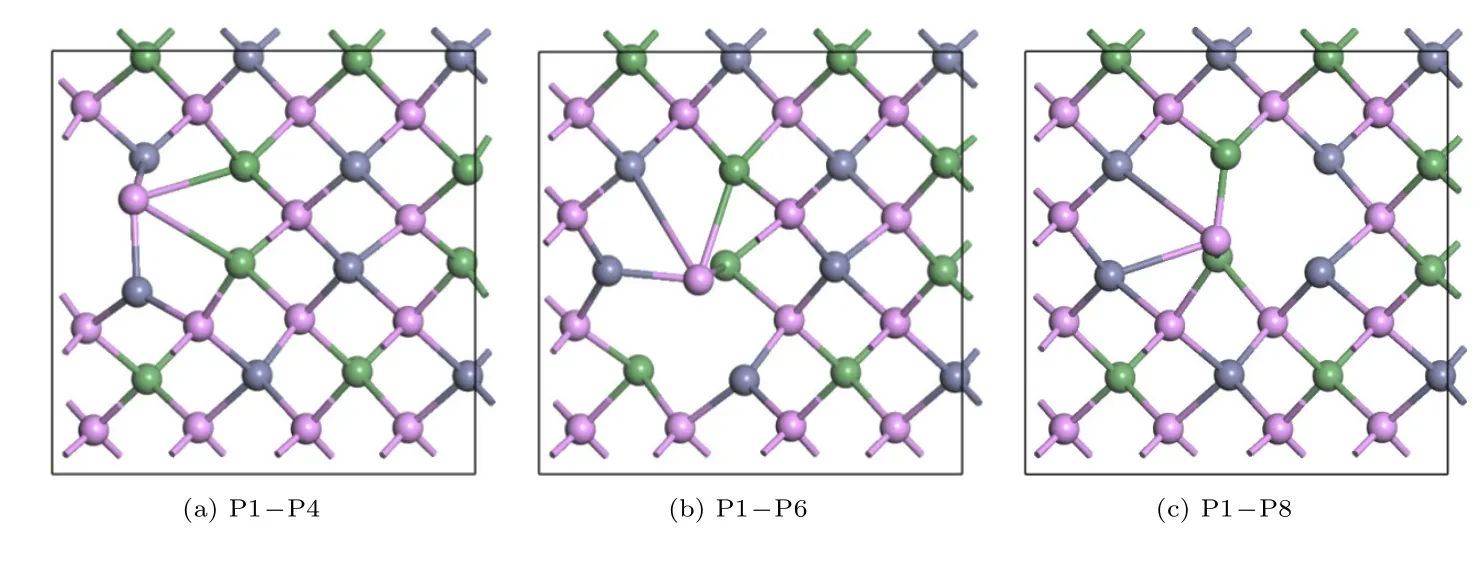
图7 (010)面P1 格点空位向P4,P6,P8 迁移的过渡态(a)P1-P4;(b)P1-P6;(c)P1-P8Fig.7.Transition states for migrations from P1 vacancy lattice to P4,P6 and P8 lattices for(010)plane:(a)P1-P4;(b)P1-P6;(c)P1-P8.

表2 (010)面P 原子间距与迁移能Table 2.P atomic spacing and migration energy for(010)plane.
3.5.2 Ge 空位迁移
采用1× 2× 2 超胞体系研究Ge 空位迁移机制,图8 给出(100)面Zn,Ge,P 原子的位置,并标记出Ge1 原子周围Ge2—Ge8 原子的位置.表3给出Ge1 原子空位向Ge2—Ge8 迁移的迁移能和原子间距.

图8 (100)面Ge 原子标记图Fig.8.Map of positions of Ge atoms for(100)plane.
从表3 可以看出,Ge1-Ge4 原子间距离最大,迁移能也最大为5.7—5.8 eV,Ge1-Ge2 原子间距离相对较小,迁移能也较小为2.2—2.4 eV 之间.通过过渡态搜索,Ge1-Ge2,Ge1-Ge3,Ge1-Ge5,Ge1-Ge6和Ge1-Ge8 分别找到对应的过渡态,Ge1-Ge4和Ge1-Ge7,没有找到过渡态,只找到中间体,而且这两组迁移能较大,均大于5 eV,表明这两组迁移不能发生.图9 给出ZnGeP2晶体(100)面Ge1 格点空位向Ge2,Ge3,Ge4,Ge7 迁移的过渡态和中间体.通过比较迁移能数据发现Ge1-Ge2正向和逆向迁移,迁移能都是最小的,因此在ZnGeP2晶体(100)面Ge 空位的迁移方向为Ge1空位向Ge2 迁移或Ge2 空位向Ge1 迁移.

图9 (100)面Ge1 格点空位向Ge2,Ge3,Ge4,Ge7 迁移的过渡态和中间体(a)Ge1-Ge2 过渡态;(b)Ge1-Ge3 过渡态;(c)Ge1-Ge4 中间体;(d)Ge1-Ge7 中间体Fig.9.Transition states and intermediate products for migrations from Ge1 vacancy lattice to Ge2,Ge3,Ge4 and Ge7 lattices for(100)plane:(a)Transition state of Ge1-Ge2;(b)transition state of Ge1-Ge3;(c)intermediate product of Ge1-Ge4;(d)intermediate product of Ge1-Ge7.
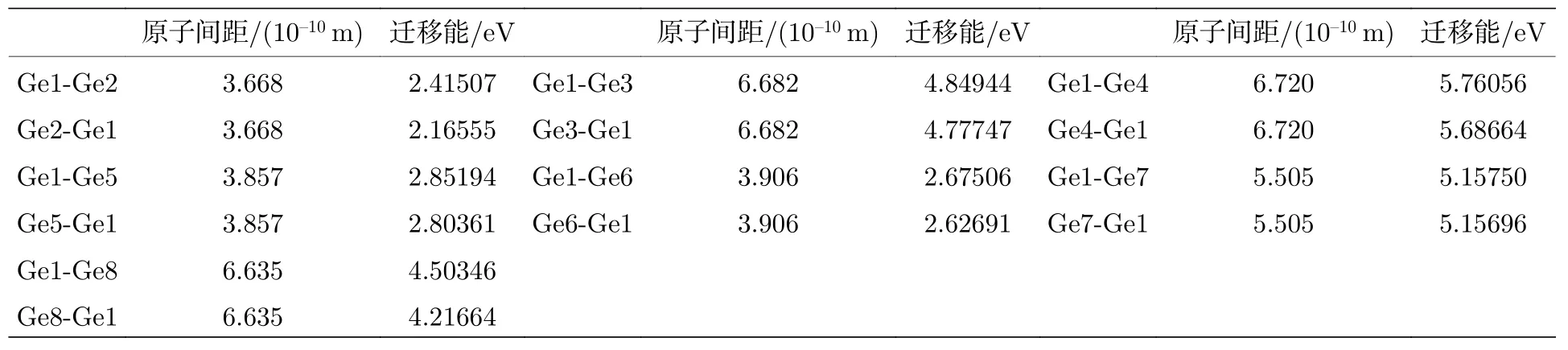
表3 (100)面Ge 原子间距与迁移能Table 3.Ge atomic spacing and migration energy for(100)plane.
3.5.3 Zn 空位迁移
Zn 空位迁移机制研究采用1×2×2 超胞体系,图10 给出(100)面Zn,Ge,P 原子的位置,并标记出Zn1 原子周围Zn2-Zn8 原子的位置.表4给出Zn1 原子空位向Zn2—Zn6 迁移的迁移能和原子间距.由于Zn1-Zn7和Zn1-Zn8 原子间距离较大,搜索不到过渡态和中间体,因此没有计算迁移能.

图10 (100)面Zn 原子标记图Fig.10.Map of positions of Zn atoms for(100)plane.
根据表4 数据,Zn1-Zn5和Zn1-Zn6 原子间距离较大,迁移能也较大,并且在搜索过渡态时,没有找到对应的过渡态,只找到中间体,表明这两组迁移不能发生.Zn1-Zn2,Zn1-Zn3和Zn1-Zn4原子间距离相近,均有相应的过渡态,但Zn1-Zn2 迁移能更小一些为1.8 eV,3 组原子空位正向迁移能均小于逆向迁移能,表明Zn 空位的迁移方向为Zn1 空位可以向Zn2,Zn3和Zn4 迁移,3 组原子空位迁移能大小顺序为Zn1-Zn3 >Zn1-Zn4 >Zn1-Zn2.图11 给出ZnGeP2晶体(100)面Zn1 格点空位向Zn2和Zn3 迁移的过渡态.通过比较迁移能数据发现Zn1-Zn2 正向和反向迁移,迁移能都是最小的,因此在ZnGeP2晶体(100)面Zn 空位的迁移方向为Zn1 空位向Zn2 迁移或Zn2 空位向Zn1 迁移.

图11 (100)面Zn1 格点空位向Zn2和Zn3 迁移的过渡态(a)Zn1-Zn2;(b)Zn1-Zn3Fig.11.Transition states for migrations from Zn1 vacancy lattice to Zn2 and Zn3 lattices for(100)plane:(a)Zn1-Zn2;(b)Zn1-Zn3.

表4 (100)面Zn 原子间距与迁移能Table 4.Zn atomic spacing and migration energy for(100)plane.
4 结论
本论文通过研究VP,VGe,VZn,ZnGe,GeZn+VZn和GeZn六种缺陷的形成能,体积膨胀率,给出了ZnGeP2晶体缺陷结构与光学性能的关系.计算了ZnGeP2晶体中VP,VGe,VZn向邻近格点迁移过程中的迁移能,并找到迁移过程中的过渡态或中间体.


2)晶体的体积和膨胀率与缺陷形成能负相关,即晶体体积膨胀率越大,缺陷形成能越低.P 空位缺陷和反位缺陷晶胞的Zn—P 键和Ge—P 键均比无缺陷晶胞的Ge—P 键和Zn—P 键键长增大,导致晶格变形,但P 空位缺陷的晶胞体积收缩,反位缺陷晶胞体积增大.反位缺陷中Zn的电荷显著增大,Ge的电荷明显降低,P的电荷无明显变化.
3)缺陷超晶胞的差分电荷密度分析显示GeZn和VZn+GeZn两种缺陷结构的Ge-Ge和Ge-Zn 原子间电子云密度增大.当VZn和VGe空位缺陷与GeZn和ZnGe反位缺陷形成联合缺陷后,空位缺陷格点处电子云密度增强.
4)10 K 时ZnGeP2晶体6 种缺陷的吸收谱表明VGe,VZn,ZnGe和GeZn4 种缺陷结构在0.6—2.5 µm 有较明显吸收,而VP和GeZn+VZn缺陷结构在此波段吸收较少,与实验测试结果相符合.
5)根据3 种空位缺陷迁移能分析,VZn的迁移能最低,VGe迁移能最高.VGe和VZn在迁移过程中原子间距离是最重要因素,原子间距离大迁移能高,反之亦然.VP在迁移过程中空间位阻是重要因素,这可能与ZnGeP2晶体中P 原子半径小且密集程度大有关.
