家蚕茧层率相关基因筛选及其功能分析
2022-12-02任晓晓孙运朋杨万军罗朝斌
任晓晓,孙运朋,卿 卓,杨万军,罗朝斌
(贵州省农业科学院蚕业研究所,贵州 贵阳 550006)
0 引言
【研究意义】蚕丝是家蚕利用最广泛的产物,不仅可用于制作丝绸制品,而且随着丝素蛋白和丝胶蛋白新用途的开发,蚕丝在生物医学、工程材料、食品和化妆品领域也具有较好的应用前景[1]。茧层率是反映家蚕对丝素蛋白、丝胶蛋白合成能力的重要性状,因此挖掘茧层率关键基因,对家蚕分子遗传育种改良和提高家蚕饲养经济效益具有重要意义[2]。【前人研究进展】家蚕饲养至今,茧层率性状经过两次较大的改良,茧层率提高至20%左右,甚至部分高茧层率品种达到30%[3]。但由于家蚕茧层率性状与其生命力、生殖力或抵抗力具有一定的相关关系,部分高茧层率家蚕品种未能在生产上推广应用。采用分子遗传育种手段定向改良茧质性状,是近年来家蚕育种工作的重点[4]。鲁成等[5]、李斌等[6]、司马杨虎等[7]利用AFLP(Amplified fragment length polymorphism,AFLP)标记构建了家蚕分子遗传图谱;侯成香等[8]、Li等[9]构建了SSR(Simple sequence repeat,SSR)和SNP(Single nucleotide polymorphism,SNP)分子遗传图谱,使家蚕茧层率QTL(Quantitative trait loci,QTL)定位研究取得了一定进展。混合群体分离分析(Bulked segregant analysis,BSA)与二代测序相结合的方法(BSA-Seq)是利用目标性状存在差异的2个亲本构建子代分离群体,在分离群体中选取极端性状个体构建DNA混池,通过高通量测序、分子标记挖掘等技术,对目标性状进行基因定位的有效手段[10-13]。BSA-Seq相比构建分子遗传图谱具有测序群体小、成本和测序要求低等优势,已经在油料、水果等作物的农艺性状QTL定位上广泛应用[14-18]。柳海东等[16]利用甘蓝型油菜早花亲本No.4512和晚花亲本No.5246构建成DH系,从中选取早花和晚花单株各20个,利用BSA-seq方法对早花位点cqDTFC8进行定位分析,将早花位点定位在C8染色体上的1.3~6.0 Mb,筛选出2个与光周期调控有关的基因,命名为BnaC08g04860D和BnaC08g04960D。尹明智等[17]利用BSA-Seq法对油菜胞质雄性不育恢复基因进行了定位分析,将恢复基因定位在C09染色体的0~880 000 bp区域,共筛选出16个基因与育性恢复有关。欧点点等[18]利用甜瓜黄皮材料和绿皮材料构建了F2分离群体,利用BSA-Seq法将皮色相关基因定位在第4染色体的0.02~5.7 Mb区间和第10染色体的0.08~9.54 Mb区间。BSA-Seq在家蚕茧质性状主效基因定位上也有应用的报道,Li等[2]基于BSA-Seq方法将与丝产量连锁的标记定位在第11、22和23号染色体上,结合候选基因表达模式、丝腺表达分析,筛选出1个调控家蚕茧层量的关键基因,命名为BmRPL18。【本研究切入点】目前有关茧层率相关QTLs的功能验证还有待进一步研究。【拟解决的关键问题】本研究以多丝量家蚕和中丝量家蚕品种为亲本,以构建的BC1群体为研究对象,采用BSA-seq方法对茧层率相关基因进行定位分析,进一步挖掘茧层率关键基因,为高茧层率关键基因克隆及家蚕经济性状分子遗传育种实践提供理论依据。
1 材料与方法
1.1 试验材料
重测序材料选取多丝量家蚕品种菁松(高茧层率)和中丝量家蚕品种芙蓉(中茧层率)构建的BC1群体,亲本材料由贵州省蚕业研究所提供,均经多代自交,背景纯合。2019年春,在贵州遵义以菁松、芙蓉杂交获得F1代。2019年夏,在贵州贵阳饲养F1代,并以F1代雄蛾回交芙蓉,获得BC1分离群体 芙蓉×(芙蓉×菁松) 。2020年春,在遵义基地同室、同条件饲养亲本和BC1群体。
1.2 茧质性状调查
在上蔟结茧的第7天采集亲本和BC1群体的蚕茧,削茧以鉴别雌、雄蛹。因家蚕茧质性状在雌、雄个体间差异显著,为排除试验误差,本试验在亲本中随机选择雌、雄茧各20粒,在BC1群体中选择所有雄性个体蚕茧(BC1M),调查记录个体的全茧量、茧层量,计算个体茧层率。
1.3 基因组DNA提取及混池构建
根据BC1M群体的茧层率调查结果,从中选择高茧层率和低茧层率极端分离材料各30个,同时取芙蓉、菁松2个亲本,运用DNA提取试剂盒提取样本基因组DNA,用1%的琼脂糖凝胶电泳检测DNA的完整性,用NanoDrop2000检测DNA样品的纯度,运用PicoGreen检测DNA的浓度。质检合格后将30个高茧层率样本DNA和30个低茧层率样本DNA分别等量混合,构建高茧层率子代DNA混池(H池)和低茧层率子代DNA混池(L池),并分别提取2个亲本的基因组DNA构建2个亲本池。
1.4 混池基因组重测序及变异位点检测
将基因组DNA委托给上海美吉生物医药科技有限公司进行建库测序,测序平台为Illumina Novaseq 6000。亲本池测序深度为10×,后代混池测序深度为20×。测序数据(Raw data)下机后进行质量控制,过滤得到高质量的Clean data。利用BWA软件将Clean data比对家蚕P50参考基因组序列(http://silkbase.ab.a.u-tokyo.ac.jp/cgi-bin/download.cgi), 获得序列的位置归属BAM文件。利用GATK的Best Practices流程对BAM文件进行校正,并进行SNP标记的变异检测,利用SnpEff软件和参考基因组的基因预测信息进行SNP的功能注释,筛选可能影响茧层率的变异位点。
1.5 连锁分析
过滤掉两子代混池间基因型一致的位点、有多个基因型的位点、read支持度小于4的SNP位点。以高茧层率亲本菁松为参考,基于过滤和筛选获得的SNP位点,对H池、L池中每个位点的SNP-index值进行计算;以0.5 Mb为窗口,10 kb为步长,采用滑窗策略对子代SNP-index在染色体上的分布进行作图,并取H池与L池SNP-index的差值计算Δ(SNP-index);进行1 000次置换检验,选择99.99%置信水平为筛选阈值,将Δ(SNP-index)在置信水平之上的窗口作为候选区域。
1.6 候选区域基因的功能注释
应用BLAST软件对候选区域的编码基因进行NR(Non redundant)、GO(Gene Ontology)、KEGG(Kyoto encyclopedia of genes and genomes)、UniProt(Universal protein)、 EggNOG(Evolutionary genealogy of genes: Non-supervised orthologous groups)数据库的深度注释,对候选基因进行功能预测。
2 结果与分析
2.1 基因组重测序及变异位点检测
对30个高茧层率个体、30个低茧层率个体及2个亲本材料进行全基因组重测序,共获得原始数据量(Raw data)10.32 Gb,过滤后得到高质量Clean reads数 在 38 269 934~ 107 624 014, Q20≥97.91%,Q30≥93.56%,GC含量为 38.66%~38.96%(表1)。将样品高质量测序数据与参考基因组进行比对,平均比对率为98.86%,成功比对到基因组上的reads比例为81.29%~82.72%,平均测序深度为18.35×,平均基因组覆盖度为95.79%(1×)、88.63%(5×)(表2)。说明样本数据量足够,测序质量良好,GC分布正常,建库测序成功;质控数据与家蚕参考基因组同源性较高,比对结果正常,可用于后续变异检测与定位分析。
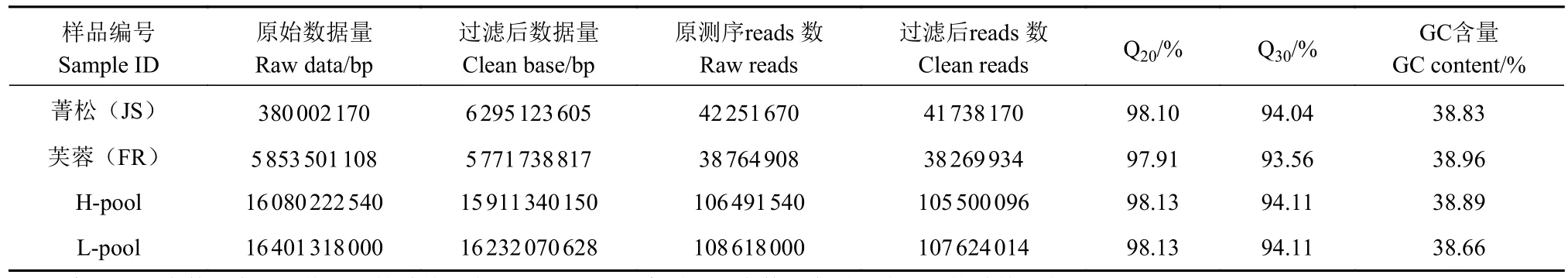
表1 测序数据质量Table 1 Statistics on quality of sequencing data
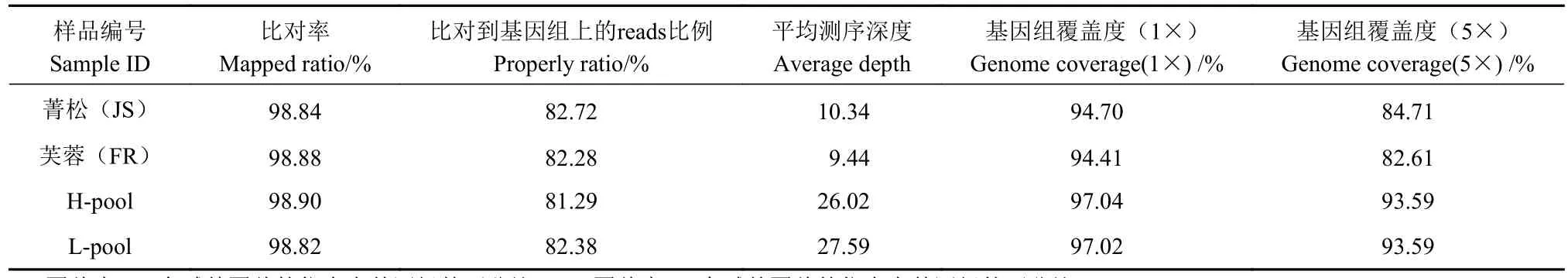
表2 质控数据与参考基因组比对情况Table 2 Matching between quality control data and reference genome
根据样品数据与家蚕参考基因组比对结果,利用GATK软件进行SNP变异位点检测,共获得26 557 646个SNP标记。利用SnpEff软件进行变异位点功能预测,结果表明SNP位点主要位于基因间区、内含子区域、基因上游和基因下游,非编码区域的多态性位点显著多于编码区。
2.2 连锁分析
根据SNP过滤原则,对变异检测获得的位点进行过滤,最终得到374 463个高质量SNP位点,使用Circos软件对变异位点在染色体上的分布密度进行可视化(图1)。根据两子代混池的SNP-index进行连锁分析,结果显示Δ(SNP-index)在置信水平之上的区域有3个,分别位于第2、4、13染色体上(图2),总长度为 1.57 Mb,共包含 67个 SNP位点、11个InDel位点、70个编码基因(表3)。
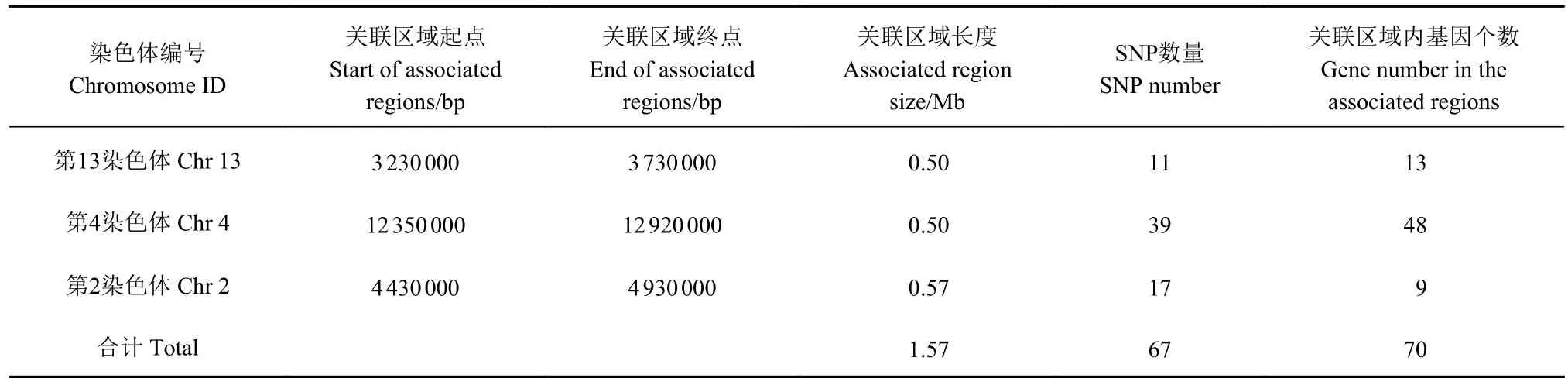
表3 关联区域信息统计Table 3 Statistics of the related genes
2.3 候选区域基因注释及候选基因筛选
利用BLAST软件对关联区域内的编码基因进行多个数据库(NR、GO、KEGG、UniProt、EggNOG)注释,结果显示:70个编码基因中有69个注释到NR数据库中,69个注释到UniProt数据库中,58个注释到GO数据库中,19个注释到KEGG通路,57个注释到EggNOG数据库。
通过对GO数据库中候选区域基因参与的生物过程(Biological process)、分子功能(Molecular function)和细胞组分(Cellular component)进行分类分析,结果表明,在生物过程中候选基因主要富集在上皮细胞发育、脑室系统发育和气管发育等;在细胞组分中,候选基因多富集于细胞核、细胞质、细胞膜;在分子功能中,候选基因富集最多的是组蛋白乙酰转移酶(图3)。
为进一步了解候选区域内基因的生物学功能,对富集到KEGG数据库中的基因进行分析,结果显示候选区域中19个基因分布于34个信号通路中,涉及新陈代谢(Metabolism)、环境信息加工(Environment information processing)、 细 胞 进 程(Cellular processes)、 遗 传 信 息 加 工 (Genetic information processing)、有机体系统(Organismal systems)(表4)。根据基因参与的代谢通路分析及文献检索,共筛选出10个与家蚕茧层率密切相关的候选基因:KWMTBOMO00853、KWMTBOMO02140、KWMTBOMO02143、KWMTBOMO02145、KWMTBOMO02146、KWMTBOMO02147、KWMTBOMO02152、KWMTBOMO07652、KWMTBOMO07653、KWMTBOMO07659,可能主要参与家蚕丝腺细胞运动、能量代谢和蛋白质合成加工。

表4 候选基因的KEGG通路分析Table 4 KEGG pathway of genes in candidate regions
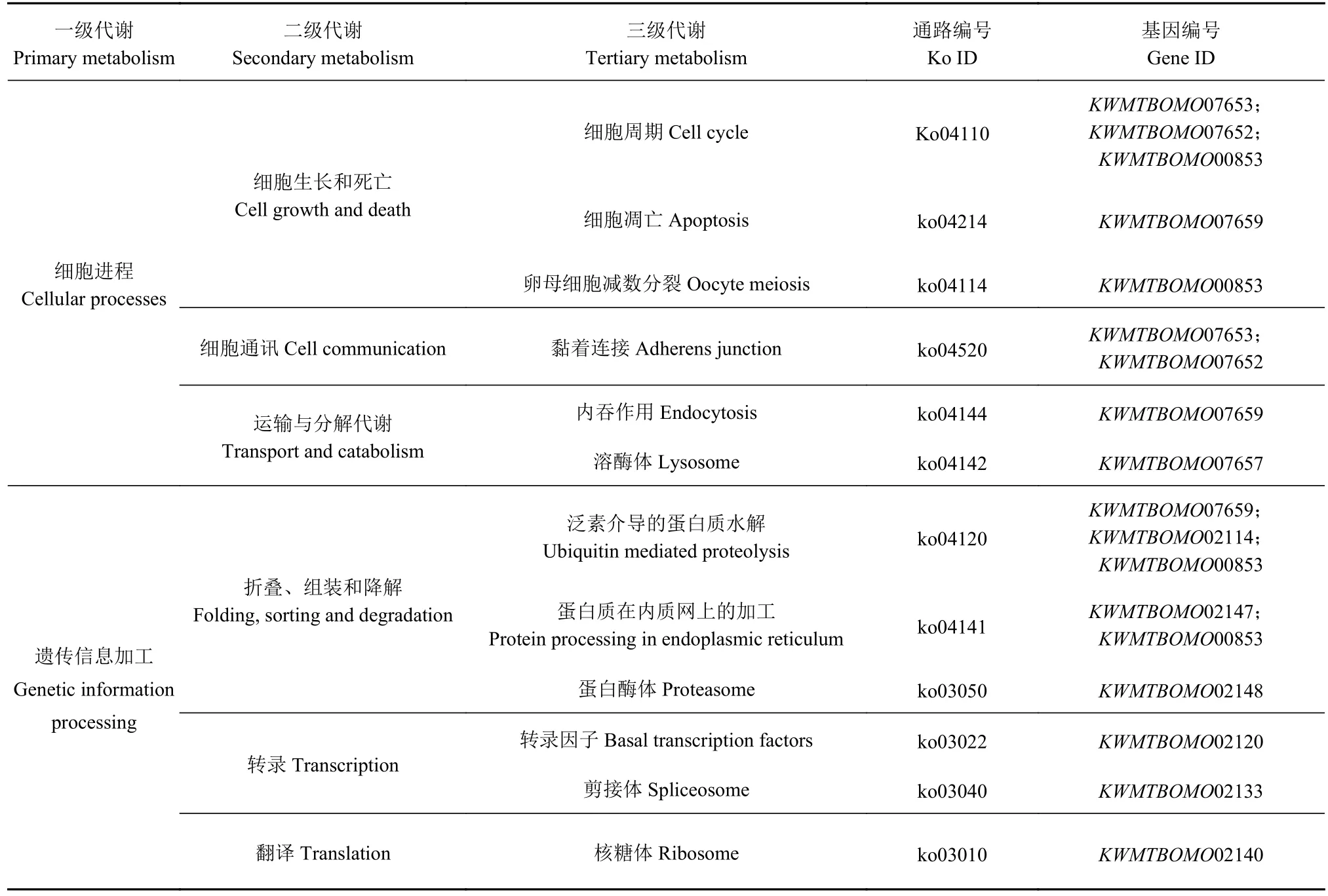
续上表
3 讨论
家蚕的部分经济性状具有性别差异,主要表现在雄蚕比雌蚕体质强健、茧层率和出丝率高,雌蚕比雄蚕全茧量和茧层量高、蛹体重大等,因此学者们推测家蚕性染色体上可能存在控制茧层率、全茧量、茧层量、蛹体重等茧质性状的基因[19-20]。Zhan等[21]以菁松和兰10为亲本构建回交一代群体(BC1M),利用SSR分子标记对家蚕部分茧质性状进行了QTL初步定位,结果在性染色体上检测到了与全茧量、茧层量和蛹体重相关的QTLs,但未检测到与茧层率相关的基因位点。侯成香等[8]在此基础上,同样以菁松和兰10为亲本配置BC1M群体,利用SSR和SNP标记构建家蚕性染色体分子遗传图谱,对茧、丝有关的QTLs进行精细扫描,但同样未扫描到与茧层率相关的QTLs。本研究以菁松和芙蓉为亲本构建BC1M群体,利用BSA-Seq分析方法对控制茧层率的基因进行定位,结果将茧层率相关的QTLs定位在第2染色体(4 430~4 930 kb)、第4染色体(12 350~12 920 kb)和第13染色体(3 230~3 730 kb),也未在性染色体上检测到与茧层率相关的基因位点。鉴于茧层率是茧层量对全茧量的比率,主要由茧层量和蛹体重决定,而前人研究表明性染色体上存在控制茧层量与蛹体重的基因,因此分析认为,茧层率与性别关联可能是由于其决定因素(茧层量、蛹体重)与性别关联。
有关茧层率的数量性状位点定位研究早有报道,鲁成等[5]利用大造和C100杂交的BC1群体,构建了AFLP分子标记连锁图谱,通过复合区间作图将茧层率相关的QTLs定位于第2、11、15连锁群上;李斌等[6]利用大造和C100的BC1群体构建AFLP分子标记连锁图谱,将茧层率相关的QTLs定位在第2、4、14、15、19、25连锁群上;司马杨虎等[7]对湘晖和872构建的F2群体构建了AFLP分子标记连锁图谱,将茧层率性状的QTLs定位在第2、11、13、18连锁群上;Zhan等[21]利用SSR分子标记对菁松和兰10的BC1M群体进行QTL位点检测,将茧层率相关QTLs定位在第18和19连锁群;Li等[9]利用菁松和兰10的BC1M群体,使用STS、SSR和SNP分子标记构建遗传图谱,结果未扫描到与茧层率相关的QTLs。Fang等[22]利用夏芳和野蚕的BC1M群体构建了SNP连锁图谱,对家蚕茧丝产量相关的QTLs进行了定位,将与茧层率有关的QTL定位在第13号染色体上。结合本研究结果可以看出,无论采用相同或不同的亲本材料、作图群体和分子标记,家蚕茧层率相关基因的定位结果均有差异,定位位置重复性较差。分析认为茧层率相关基因定位重复性差与其遗传基础复杂有关,即可能存在多个家蚕茧层率性状关键基因位点,且其效应有差异。本研究筛选出了10个与茧层率相关联的候选基因,其KEGG通路分析显示,候选基因可能参与了丝腺细胞运动、能量代谢和蛋白质合成加工过程,在一定程度上证实了茧层率相关基因调控机制的复杂性。
家蚕茧丝中的丝素蛋白在后部丝腺中合成,后部丝腺细胞为核内复制方式,即细胞周期只有DNA复制时期(S期)和细胞间期(G期),而不具有分裂期(M期)和胞质分裂过程[23]。细胞周期蛋白E(Cycline-E,CycE)、细胞周期蛋白依赖激酶2(Cyclin-dependent kinases 2,CDK2)是S期的主要调节因子,与其他核内周期调节者构成复杂的调控网络,精密协作保证核内周期的顺利完成[24]。前人研究发现,果蝇Ras信号(Ras85D)可能通过调控CycE、CDK2等核内周期调节因子,参与调控细胞的存活、生长等生理过程。Caldwell等[25]通过激活果蝇前胸腺细胞(由核内复制细胞组成)中的Ras信号,使前胸腺细胞体积明显增大。Ma等[26]利用GAL4/UAS技术在家蚕后部丝腺中特异性过表达Ras1CA激活了Ras信号,促进了后部丝腺细胞和细胞核体积增大,全茧量有所增加。马倩等[23]基于RNASeq技术比较了野生型与Ras1CA过表达转基因家蚕后部丝腺组织中的差异基因,结果发现Ras信号可使细胞周期依赖蛋白激酶(CDK)、细胞周期蛋白(Cycline)和DP-1,2转录因子等编码基因上调,进而调控细胞周期通路,促进后部丝腺组织细胞核内复制,使丝腺组织增大,其认为在此过程中丝素蛋白编码基因的拷贝数也大量增加,可能有利于丝蛋白的合成。本研究通过BSA-Seq方法定位到10个与茧层率性状密切相关的候选基因,其中KWMTBOMO02152直接参与了Ras信号通路;KWMTBOMO00853、KWMTBOMO07652和KWMTBOMO07653参与了细胞周期通路;KWMTBOMO02143、KWMTBOMO02145和KWMTBOMO07659参与了丝裂原活化蛋白激酶(Mitogen-activated protein kinase,MAPK)信号通路,该通路通过级联方式将细胞外信号逐级扩大并传导到细胞或细胞核内,进而调控细胞周期的运行和基因表达,也是Ras信号通路和细胞周期通路的重要组成部分;KWMTBOMO02146参与了胰岛素信号通路和钙离子信号通路,其中胰岛素信号通路参与调控能量代谢、细胞生长及蛋白质合成过程,钙离子信号通路参与Ras信号通路;KWMTBOMO02140参与了核糖体信号通路,与丝素蛋白的合成密切相关;KWMTBOMO00853、KWMTBOMO02147参与了蛋白质在内质网上的加工。这些基因是否是调控家蚕茧层率的关键基因及其调控机制将是本课题组下一步的研究重点。
4 结论
运用BSA-seq方法成功地在家蚕第2染色体、第4染色体和第13 染色体上定位到与茧层率关联的区域;通过KEGG功能注释及相关文献检索,筛选到10个可能与茧层率密切相关的基因,参与了Ras信号通路、细胞周期通路、MAPK信号通路、胰岛素信号通路、钙离子信号通路、核糖体信号通路和蛋白质在内质网上的加工过程。研究结果为高茧层率相关调控基因的精细定位奠定了基础。
