NLRP3炎症小体激活促进肝星状细胞活化的机制
2022-11-16银思涵孙克伟
彭 憬,袁 维,银思涵,孙克伟
湖南中医药大学第一附属医院 肝病科,长沙 410007
肝纤维化是一系列慢性肝损伤的病理生理结果,是一个涉及实质细胞(肝细胞)、肝星状细胞(hepatic stellate cell,HSC)、肝窦内皮细胞以及常驻和浸润免疫细胞之间相互作用的动态过程,细胞外基质(extracellular matrix,ECM)的过度积累则是其最突出的特征[1]。HSC位于肝窦内皮细胞和肝细胞之间的窦周隙,主要在胞浆的脂滴中储存类视黄醇(维生素A)。激活后的HSC失去储存类视黄醇的能力,开始增殖、收缩和分泌ECM主要成分胶原Ⅰ和Ⅲ[2]。肝窦内皮细胞上的窗孔会被HSC形成的基底膜封闭,造成肝窦毛细血管化,影响血窦与肝细胞之间物质交换[3]。近年研究发现,NOD样受体蛋白3(NOD-like receptor protein 3,NLRP3)炎症小体可在肝细胞[4]、HSC[5]、肝巨噬细胞[6]等多种细胞内表达,除介导HSC的激活外,还通过释放的下游炎症因子和引起细胞焦亡放大肝脏内炎症反应,形成恶性循环加重肝脏损伤和纤维化。
NLRP3是三元蛋白质,包含一个氨基端吡啉结构域(PYD),一个中央NACHT结构域(NAIP、CIITA、HETE和TP1)和一个羧基端富含亮氨酸重复序列(LRR)结构域。在炎症小体复合物组装过程中,作为接头蛋白的凋亡相关斑点样蛋白(apoptosis-associated speck-like protein,ASC)N端通过PYD-PYD与NLRP3连接。ASC的C端有一个caspase募集结构域(CARD),通过CARD-CARD相互作用与procaspase-1结合,促进后者二聚化和激活。随后以caspase-1依赖的方式,释放炎性介质IL-1β和IL-18(图1)。NLRP3炎症小体作为胞浆内蛋白感受器,目前认为它的激活分两步:首先由LPS、TNFα等启动NLRP3和pro-IL-1β表达,接着被K+外流、Ca2+内流、线粒体功能紊乱、细胞内活性氧(reactive oxygen species,ROS)等胞内损伤信号激活[7-8]。影响HSC活化的因素如IL-1、TNF、ROS等[2]也能激活NLRP3炎症小体,提示后者可能参与了HSC活化。基于NLRP3 炎症小体激活在肝纤维化中的重要作用,NLRP3有望成为临床治疗的新靶点。
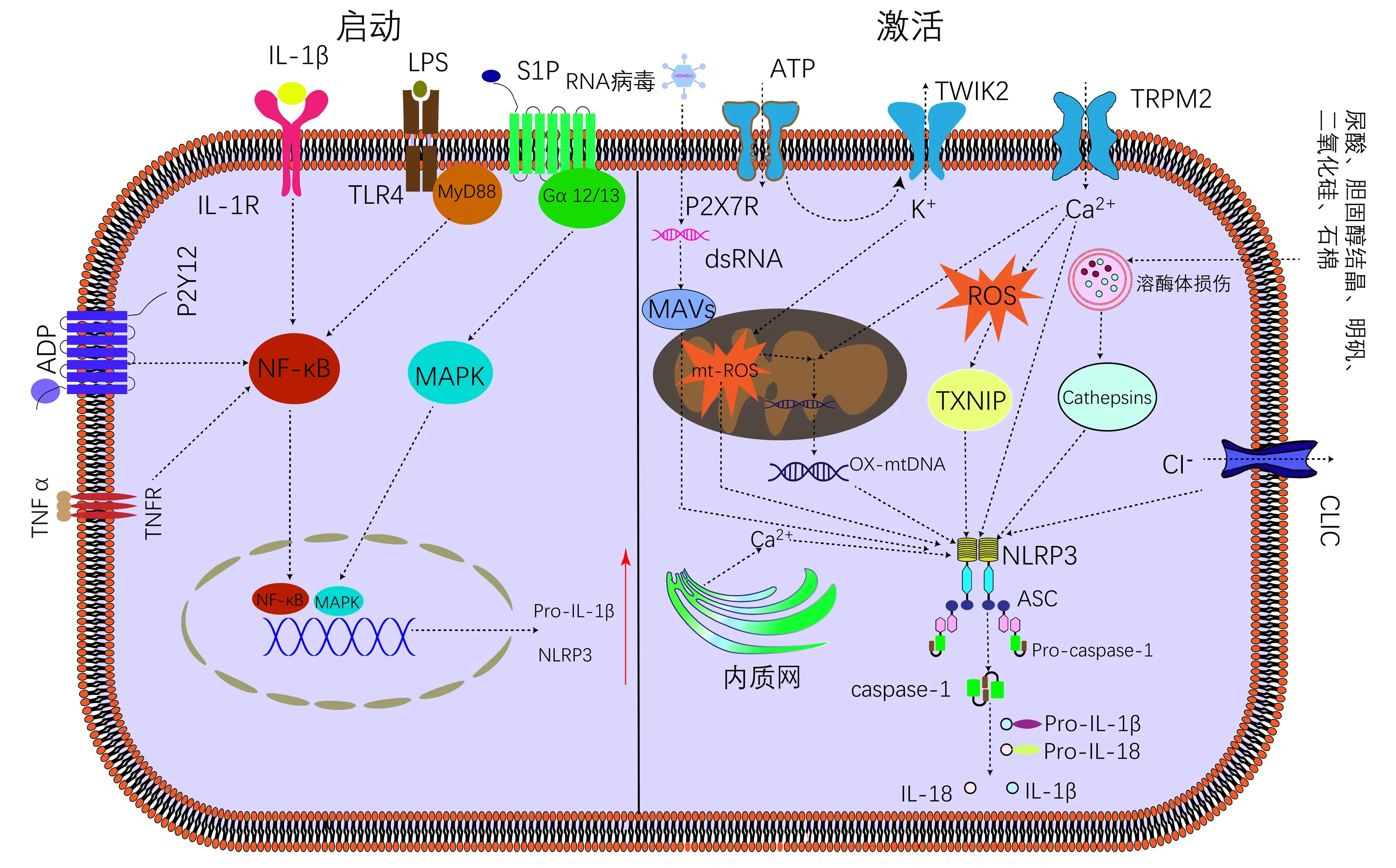
注:微生物组成部分和内源性损伤因子提供启动信号,导致关键转录因子NF-κB激活,随后上调NLRP3和pro-IL-1β表达。细胞外ATP、K+外流、Ca2+内流、ROS的产生、溶酶体损伤、颗粒物质等激活NLRP3炎症小体。dsRNA,双链RNA;TLR4,Toll样受体4;ROS,活性氧;Ox-mtDNA,氧化线粒体DNA;TXNIP,硫氧还蛋白互作蛋白;TWIK2,弱内向整流二孔K+通道2;TRPM2,瞬时受体电位M2通道;P2X7R,嘌呤能离子通道型受体7;MAVS,线粒体抗病毒信号蛋白;CLIC,胞内氯离子通道。
1 介导HSC内不同通路
活化的HSC是肝纤维化过程的关键细胞,研究发现NLRP3炎症小体可通过多条通路激活HSC(图2),促进其α平滑肌肌动蛋白(α-SMA)和胶原等激活标志的表达,引起肝纤维化。
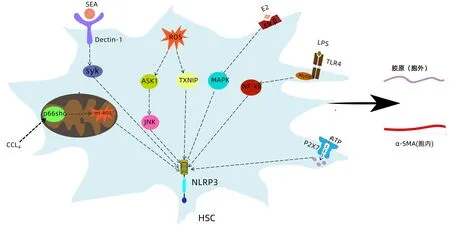
注:NF-κB,核因子κB;MyD88,髓样分化因子88;MAPK,丝裂原活化蛋白激酶;Dectin-1,树突状细胞相关C型凝集素-1;ERβ,雌激素受体β;ASK1,凋亡信号调节激酶1;Syk脾酪氨酸激酶;SEA,日本血吸虫可溶性虫卵抗原;p66Shc,66 kDa原癌基因Src胶原同源物(Shc)衔接蛋白。
1.1 TLR4/MyD88/NF-κB/NLRP3信号通路 用LPS刺激HSC上TLR4,能活化下游NF-κB炎症信号通路[9],而NF-κB作为关键转录因子,能提供启动信号,上调NLRP3和pro-IL-1β表达。棕榈酸(PA)通过刺激TLR4/NF-κB信号通路,激活HSC中的NLRP3炎症小体并向纤维化表型转变。另外被此信号通路激活的NLRP3炎症小体,加剧了高脂肪饮食喂养的非酒精性脂肪性肝炎(nonalcoholic steatohepatitis,NASH)大鼠向肝纤维化的转变[10]。血管紧张素Ⅱ(Ang Ⅱ)通过 TLR4/MyD88/NF-κB通路促进NLRP3和pro-IL-1β的表达,促进HSC的激活。此外,IL-1β还能协助Ang Ⅱ增加TLR4、MyD88和Ⅰ型胶原Α1(collagen type Ⅰ alpha 1,Col1a1)蛋白水平的表达[11]。TLR4模式识别受体是固有免疫识别各种病原体的重要组成部分,能识别多种内源性和外源性配体,刺激表达在HSC上的TLR4使其活化,可能在多种肝脏疾病纤维化进展中发挥重要作用。
1.2 嘌呤能离子通道型受体7(P2X7R)/NLRP3信号通路 P2X7R能被受损细胞释放的ATP激活,在NLRP3炎症小体激活的第二信号中起关键作用。在LPS/ATP双重信号刺激下,LX-2 细胞(人肝星形细胞系)中NLRP3炎症小体各组成部分显著上调,用选择性P2X7R拮抗剂A438079或P2RX7 siRNA能抑制HSC中NLRP3炎症小体的表达和ECM的合成与沉积[12]。P2X7R的过表达还降低了LX-2细胞激活的阈值[13]。乙醇主要在肝细胞中代谢,但过量的乙醇积累能损伤肝细胞,使其释放ATP[14]。一项研究[15]显示在慢性酒精性肝纤维化和急性酒精性肝损伤小鼠模型中,P2X7R及相关基因的表达均上调,HSC内此通路的激活还可以使其自分泌IL-6、TNFα等炎症因子以及激活静止HSC成为肌成纤维细胞的TGFβ。这显示在酒精性肝病中,肝细胞与HSC之间的相互作用除能通过P2X7R直接激活HSC外,还可以介导炎症,在纤维化的发展中具有重要作用。
1.3 硫氧还蛋白互作蛋白(TXNIP)/NLRP3信号通路 在静息状态下,TXNIP与其配体结合并使其处于非活性状态。当细胞氧化应激时,被诱导产生的ROS触发TXNIP与硫氧还原蛋白解离,结合并激活NLRP3炎症小体[16]。TXNIP的敲低抑制HSC激活,而TXNIP过表达则增强HSC激活,并且这种方式是通过HSC内氧化应激调控的[17]。在日本血吸虫诱导的肝脏肉芽肿和肝纤维化小鼠模型中,肝脏组织中TXNIP的表达水平增加,并与NLRP3炎症小体相互作用,促进HSC α-SMA和胶原Ⅰ和Ⅲ表达增加[18]。TXNIP与HSC内氧化应激水平密切相关,许多有害刺激都能引起细胞氧化应激,TXNIP作为广泛表达的蛋白,研究其在HSC激活中作用的文献较少,有待进一步深入。
1.4 p66Shc/mito-ROS/NLRP3信号通路 p66Shc(Shc的66 kDa蛋白亚型)是线粒体活性氧(mito-ROS)产生的主要调节因子,是肝细胞氧化应激的关键介质。应激刺激后,在胞浆中激活并磷酸化的p66Shc转移到线粒体膜间隙,结合并氧化细胞色素c产生大量的mito-ROS[19]。p66Shc基因的敲低使HSC超氧化物歧化酶表达和活性显著增加,以及H2O2、mito-ROS水平和线粒体细胞色素c释放急剧减少,维持线粒体膜正常电位,改善TGFβ1引起的氧化应激,此外NLRP3炎症小体复合物组成部分表达也下调。p66Shc过度表达使HSC中激活标志物如结缔组织生长因子(CTGF)、α-SMA、Col1a1和基质金属蛋白酶组织抑制因子(TIMP1)表达增加,通过NLRP3的敲低则会减弱这种效果[20]。mito-ROS占细胞内ROS绝大部分来源,因此在氧化应激中p66Shc可能通过促进mito-ROS形成介导HSC活化。
1.5 E2/雌激素受体β(ERβ)/MAPK/NLRP3信号通路 ERβ激动剂二芳基丙腈(DPN)能抑制HSC活化,以及血小板衍生生长因子诱导的HSC增殖[21]。TGFβ能减少ERβ在HSC细胞膜上的表达,并且ERβ的选择性抑制剂四氯乙烯(THC)增强了NLRP3的激活[22]。在H2O2诱导的HSC-T6细胞氧化应激中,三个MAPK蛋白(p-ERK、p-JNK和p-p38)磷酸化显著增加,雌二醇(E2)处理组能下调它们的表达,THC则能完全抑制E2的作用,而ERα选择性抑制剂甲基哌啶吡唑(MPP)却无此效果[23]。综上所述,说明E2主要通过ERβ发挥抑制肝纤维化的作用。这可能也解释了在绝经前,同龄的女性肝纤维化程度较轻,而绝经后这一保护作用却消失[24]。
1.6 Dectin-1/Syk/NLRP3信号通路 树突状细胞相关C型凝集素-1(DC-associated C-type lectin-1,Dectin-1)是细胞膜蛋白,可依赖其下游信号脾酪氨酸激酶(spleen tyrosine kinase,Syk)诱导ROS的产生,触发NLRP3炎症小体组装[25]。用Dectin-1阻滞剂预处理小鼠HSC,阻止了日本血吸虫可溶性虫卵抗原(SEA)诱导的caspase-1和IL-1β的产生。Syk基因的沉默则抑制了NLRP3和caspase-1在HSC中的聚集[26],考虑到Syk能使ASC第146 和第187位酪氨酸残基磷酸化,这一过程能增强ASC寡聚化并招募procaspase-1[27],推测上述实验结果可能是Syk可与作为接头的ASC直接结合,促进炎症小体复合物形成有关。NLRP3基因的沉默显著抑制SEA刺激小鼠HSC向纤维化表型改变[26],提示Syk可通过ROS的产生和直接促进炎症小体复合物的形成,在此通路中发挥关键作用。
1.7 ASK1/JNK/NLRP3信号通路 ASK1能被ROS、LPS、细胞因子等各种刺激活化,随后磷酸化JNK[28],而p-JNK可以使NLRP3第194位丝氨酸磷酸化(人是198位),促进NLRP3炎症小体的自结合和激活[29]。ASK1/JNK通路能促进HSC增殖和活化,加重大鼠肝纤维化和炎症反应[28]。在他莫昔芬诱导激活的NLRP3突变体敲入(Nlrp3-KI)小鼠,口服ASK1抑制剂GS-444217能降低肝组织磷酸化ASK1、c-Jun的表达,抑制肝细胞的死亡和肝纤维化。Nlrp3-KI小鼠分离的Kupffer细胞中促炎基因TNFα、IL-1β及HSC中促纤维化基因Col1a1、肌动蛋白α2的表达增强,而ASK1的抑制能使上述基因的表达减弱[30]。在一项多中心二期临床实验,ASK1选择性抑制剂selonsertib能改善NASH患者2期或3期肝纤维化和肝小叶炎症[31],显示此通路的激活与肝脏炎症反应和纤维化密切相关。
2 促进炎症微环境形成
肝脏炎症微环境由Kupffer细胞、浸润的巨噬细胞、T淋巴细胞、树突状细胞和中性粒细胞及其分泌的炎性介质构成。而其中巨噬细胞在促进HSC活化中至关重要,激活后不仅能分泌炎症因子[32],作为CXCL2的主要来源,还能募集中性粒细胞[33]。而NLRP3炎症小体的激活能促进肝脏炎症微环境的形成,进而使HSC活化。
2.1 肝脏巨噬细胞 与HSC中NLRP3炎症小体的激活会诱导其向纤维化表型转变不同,巨噬细胞中NLRP3炎症小体的激活可使大量炎症因子释放到胞外,进而促进HSC活化。在胆石酸(LCA)+Nigericin刺激下,Kupffer细胞内ROS的增加能激活NLRP3炎症小体,使得M1表型标志Tnf、iNos表达增加,M2表型标志Arg1表达下调[34]。而应用NLRP3的抑制剂MCC950,使肝脏极化巨噬细胞向M2型转变[35],以上实验提示在巨噬细胞向促炎性表型转换中,NLRP3炎症小体的激活起重要作用。M1型巨噬细胞分泌的TNFα[36]、IL-6[37]等炎症因子可促进HSC的激活,而分泌的IL-1β作用于HSC上的P2X7R,可以使其维持在激活状态[13]。不同于前述巨噬细胞内NLRP3炎症小体的激活促进HSC活化,巨噬细胞分泌的CXCL2可以作用于LX-2细胞上受体CXCR2,介导HSC内NLRP3炎症小体激活使其活化[38]。
2.2 中性粒细胞 NLRP3炎症小体在转基因小鼠体内的激活,可以上调中性粒细胞关键趋化因子CXCL1(20倍)和CXCL2(30倍)的mRNA表达[39]。NLRP3基因敲除的小鼠肝脏髓过氧化物酶(MPO)+细胞(中性粒细胞)渗入减少[34],表明NLRP3炎症小体的激活可募集中性粒细胞到肝脏,加重炎症反应。HSC可被募集到肝脏的中性粒细胞,产生的ROS激活,并分泌粒细胞-巨噬细胞集落刺激因子和IL-15,延长中性粒细胞的生存[40],提示中性粒细胞与HSC之间存在正反馈回路,促进肝脏损伤和纤维化。
2.3 IL-1β和IL-18 NLRP3炎症小体下游信号IL-1β和IL-18也可以介导炎症反应,但IL-1β还能与HSC相互作用促进其激活。IL-1β介导TGFβ以自分泌方式在LX-2细胞表达[41],造成ECM沉积和NLRP3炎症小体活化的第二信号如P2X7R激活及Ca2+内流增加[13],显示存在正反馈环路促进HSC的激活。用IL-1β受体阻滞剂rIL-1Ra预处理HSC-T6细胞,E.coli RNA诱导产生的TGFβ1的分泌明显降低,细胞内α-SMA、COL1A1和TIMP-1的mRNA表达被显著抑制[42]。上述研究发现IL-1β能影响HSC的TGFβ分泌和活化,其机制在于IL-1β能活化NF-κB亚基p65和转录激活蛋白-1(activator protein-1,AP-1)亚基c-Jun,分别结合到TGFβ启动子κB3和AP-1-3位点,提高组蛋白H4和H3乙酰化水平,募集RNA聚合酶Ⅱ启动TGFβ基因转录[43]。此外IL-1β还能通过JNK/AP-1通路诱导体外培养的HSC增殖,但在小鼠体内却不明显[44-45]。
3 总结与展望
NLRP3炎症小体在多种肝脏疾病中都可表达,与疾病的严重程度相关[4,46],可作为肝组织炎症和损伤的检测指标。NLRP3炎症小体通过HSC内氧化应激、炎性信号等,促进由巨噬细胞、中性粒细胞、IL-1β等构成的炎症微环境的形成,从而形成错综复杂的网络使HSC活化。
尽管上述众多研究表明NLRP3与HSC的活化相关,但研制针对肝纤维化的NLRP3抑制剂较少,例如NT-0167(MCC950衍生物)应用于肝纤维化仍处于临床前研究,疗效尚不清楚[47]。MCC950能非竞争性抑制碳酸酐酶2活性,造成脱靶[48],这也限制了MCC950及其衍生物在临床上的应用。由于NLRP3炎性小体有复杂的信号级联放大效应,可以使用多种靶点来抑制NLRP3炎症小体。在个案报道中,用秋水仙碱常规治疗地中海热的基础上联合IL-1β中和抗体canakinumab,可使进展为NASH的患者转氨酶降为正常,显著改善肝纤维化[49],显示了积极的抗肝纤维化效果。但由于对NLRP3阻滞剂作用机制的理解不足,以及这些分子的潜在脱靶效应限制了它们进一步的临床应用,选择NLRP3分子特异结构设计药物和提高对HSC的靶向性将是有意义的探索。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:彭憬、孙克伟参与起草和修改文章关键内容;袁维、银思涵对研究的思路有关键贡献。
