病毒感染与NLRP3炎性小体
2022-06-08贾朝霞刘开扬
曹 旭,贾朝霞,刘 浩,王 静,刘开扬
长期以来,由于各种原因使得机体被感染而产生炎症后导致全身多器官衰竭的问题一直困扰着临床工作者。而炎症的产生与炎性小体有密切联系。其中NLRP3炎性小体作为众多炎性小体中的一种,广泛参与多种病毒引起的炎症反应。各种病毒感染宿主细胞时,不同炎性小体的激活是宿主抗病原体感染所必需的。宿主受病原体刺激后能诱导NLRP3炎性小体的组装与活化,随后激活半胱氨酸天冬氨酸蛋白酶-1(caspase-1),处在活化状态的caspase-1诱导下游相应促炎因子的剪切、成熟与分泌,进而参与炎症的发生发展。临床上促使炎症发生的致炎因子有多种,包含生物性因子(如细菌、病毒、真菌等)、物理性因子(如高温、低温、放射性物质及紫外线等)、化学性因子(如强酸、强碱等)、异物(如碎屑、尘埃颗粒及手术缝线等)、坏死组织等[1]。其中,生物性因子中的病毒感染越来越多,如HIV感染最终导致全身免疫系统被攻击,机体进行免疫预防及免疫清除的能力明显下降,机体由于无法及时发现和清除病原体的入侵而造成严重感染以及肿瘤发生率明显增加。过去的10年,NLRP3炎性小体作为一个非常关键的先天免疫成分在协调宿主免疫稳态方面发挥的作用不容小觑。因此,深入研究病毒感染时NLRP3炎性小体的激活途径和调控机制,对于我们从根本上阻止病毒入侵、复制,进而控制炎症的发生发展至关重要。
1 NLRP3炎性小体的结构
NLRP3炎性小体是由NLRP3蛋白、凋亡相关斑点样蛋白(ASC)以及pro-caspase-1组成的复合物。其中,NLRP3蛋白由C端的亮氨酸重复序列(LRR)、N端的效应结构域以及中间段的核苷酸寡聚化结构域(NACTH)组成,且各部分都有其相应的功能,比如C端的亮氨酸重复序列能识别病原微生物及内源性的危险信号;N端的效应结构域主要包括热蛋白结构域(PYD)、胱天蛋白酶募集结构域(CARD)等,介导了信号传导过程;中间段核苷酸寡聚化结构域促使NLR寡聚化。NLRP3炎性小体中的ASC作为中间部分起到了连接NLRP3和pro-caspase-1的作用,ASC由PYD和CARD两部分组成,其中PYD能与NLRP3蛋白的PYD相互作用,CARD能招募pro-caspase-1进而介导NLRP3激活,形成具有酶活性的caspase-1[2-3]。
2 NLRP3炎性小体的激活
当细胞处于静息状态时,NLRP3炎性小体的各组成部分以及作用底物都处于一个极低水平不能被测及[4]。目前,已发现NLRP3炎性小体的激活主要包括两个过程,首先是启动过程:Toll 样识别受体(Toll-like receptor,TLR)识别相应配体,启动核因子κB (Nuclear factor kappa-B,NF-κB)介导的信号通路,诱导pro-IL-1β、pro-IL-18 和NLRP3 的表达上调。其次是激活过程:该过程由细胞外ATP、成孔毒素或颗粒物等刺激导致NLRP3炎性小体的组装、激活以及caspase-1经活化后对下游促炎因子的加工修饰并最终导致其成熟分泌的过程[5]。在激活过程中,钾离子外流、溶酶体破坏、线粒体损伤及活性氧(Reactive oxygen species,ROS)的产生是激活NLRP3炎性小体的关键因素。研究发现,病原相关分子模式以及损伤相关分子模式进入宿主体能够促进NLRP3炎性小体的活化,如ATP、病原体相关RNA、晶体或微粒、细菌毒素及某些疫苗增强剂[6]。然而这些刺激未被证实直接与NLRP3相互作用,由于其结构和化学上的差异性,怀疑它们诱导共同的细胞信号激活NLRP3炎性小体。目前,这一观点得到了许多研究者的认可[7]。也有研究认为在NLRP3炎性小体的活化过程中,宿主体内的某些激酶发挥了作用,如半胱氨酸激酶,但这一观点目前尚未达成共识[8-9]。最近的研究有了重大进展,发现了一种新的激酶与NLRP3炎性小体激活相关。NEK7,一种已知参与有丝分裂的丝氨酸激酶,其对NLRP3炎性小体的激活至关重要,能特异性的与NLRP3相互作用,且与其他炎性小体无关。
3 病毒感染对NLRP3炎性小体的激活机制
3.1 病毒RNA介导的激活 研究发现,尽管DNA病毒的种类多于RNA病毒,但目前导致人类疾病的常见病毒则是RNA病毒。最初就有研究者认为,NLRP3炎性小体能被病毒核酸RNA所激活,并介绍了激活的途径。随后病毒RNA能激活NLRP3炎性小体这一观点成为研究者们研究的热点。研究发现,将病毒RNA或其类似物注入到小鼠体内或人巨噬细胞中能激活NLRP3炎性小体[10-11]。如从轮状病毒中提取的dsRNA能激活NLRP3,从甲型流感病毒(Influenza A Virus,IAV)中提取的ssRNA也能激活NLRP3,进而启动由NLRP3介导的炎性小体的激活[12]。另有文献称,将双链RNA类似物poly(I:C)注入小鼠体内,它能刺激小鼠体内IL-1β分泌增加,且这种炎症反应是由NLRP3介导产生的[13]。尽管这些发现都与NLRP3分子有关,但病毒RNA并不是与NLRP3直接结合来启动炎性小体的活化。其激活过程为病毒RNA首先被模式识别受体(TLRs或NLRs)识别,随后通过NF-κB介导的信号途径上调pro-IL-18和pro-IL-1β和NLRP3分子的表达,然后启动NLRP3炎性小体的激活[14]。RNA活化蛋白激酶(RNA-activated protein kinase, PKR)在该激活过程中也起到了一定作用。有研究者发现,细胞受到poly(I:C)等多种刺激后,PKR能发生自磷酸化,直接与NLRP3分子相互作用,进而调节NLRP3炎性小体的活化,但目前此观点备受争议[15]。有研究者称使用PKR抑制剂2-氨基嘌呤处理小鼠后,小鼠体内炎性小体的激活程度减弱,所以相关研究者认为PKR在NLRP3炎性小体的激活中发挥了一定的作用[16]。然而,另有研究者认为PKR在炎性小体的激活中并没有发挥作用。实验对PKR缺陷的的杂合子小鼠和PKR缺陷的纯合子小鼠同时用NLRP3的激活剂进行刺激,发现在炎性小体激活方面并无差异。因此,对于PKR在NLRP3炎性小体激活中的作用需要进一步深入研究[17]。
以上是病毒RNA激活NLRP3炎性小体的主要方式。研究还发现病毒RNA激活炎性小体的另一种方式。RIG-I(retinoic acid-inducible gene I)即维甲酸诱导基因蛋白I,其属于维甲酸诱导基因I样受体家族(RLRs),同样是一种胞内模式识别受体,具有识别病毒RNA的作用。其过程为RIG-I识别病毒RNA后能与ASC和caspase-1结合,启动NLRP3炎性小体激活,该途径并无NLRP3参与介导[18]。这种激活炎性小体的方式限于某些病毒感染,如水泡性口炎病毒(Vesicular stomatitis virus,VSV)[19]。研究也发现脑心肌炎病毒(Encephalomyocarditis virus,EMCV)能借助RLRs家族中的另一个成员黑色素瘤分化相关抗原5(melanoma differentiation-associated gene 5, MDA5),作为胞内的一种模式识别受体识别病毒RNA,通过激活NF-κB途径启动依赖NLRP3的炎性小体的活化[20-21]。近期,有学者发现在正常人的支气管上皮细胞中RIG-I能与ASC及caspase-1相互作用来调节NLRP3炎性小体活化。该研究还进一步发现RIG-I能在转录水平上对NLRP3及促炎因子IL-1β的前体进行调节,提示RIG-I具有启动和激活炎性小体的双重作用[22]。然而另有研究表明,在少数病毒如风疹病毒(Rubella virus, RV)感染中,RIG-I辨别RV的RNA后,RIG-I没有直接参与炎性小体的活化,其具体的参与机制有待后续的探索[23]。
3.2 病毒感染致宿主细胞状态变化介导的激活 病毒感染过程中会引起宿主细胞状态发生一系列变化,如细胞内离子浓度异常、溶酶体破坏和线粒体功能障碍等,这些变化会被宿主感应为危险信号进而导致NLRP3炎性小体的激活。
细胞内外各种离子保持适当的梯度是维持宿主细胞稳态的前提。如静息状态下,细胞内K+浓度约是胞外K+浓度的30倍,而胞外Na+浓度约是胞内Na+浓度的15倍[24]。相反,一旦这种稳态被打破,NLRP3转录体将感知到危险信号,上调NLRP3炎性小体组分的表达,随后组装激活NLRP3炎性小体。如被熟知的K+外排能激活NLRP3炎性小体事件,在丙型肝炎病毒(Hepatitis C virus,HCV)感染巨噬细胞中得到了证实[25]。此外,离子浓度的紊乱会导致线粒体损伤和 ROS 的产生,从而增强 NLRP3 炎性小体的激活[26]。
溶酶体破坏会导致内容物组织蛋白酶B的释放,随后诱导ROS产生,刺激NLRP3炎性小体激活。如腺病毒(Adenovirus,ADV)感染诱导溶酶体破坏和组织蛋白酶B的释放,从而激活NLRP3炎性小体[27]。IAV感染通过诱导溶酶体酸化来激活 NLRP3 炎性小体[28]。此外,NLRP3 炎性体激活也需要 ROS,因为在NADPH 氧化酶抑制剂或氧清除剂 N-乙酰半胱氨酸存在的情况下观察到较低水平的 IL-1β[29]。
线粒体损伤也是NLRP3炎性小体激活的关键因素。其同样诱导ROS产生促使NLRP3炎性小体激活。一项研究报道,病毒感染后促使RIP1-RIP3复合物组装随后诱导GTP酶DRP1激活。而DRP1能转移到线粒体调节其异常功能和致使线粒体损伤[30]。另有几项研究发现,多种病毒及RNA类似物激活NLRP3炎性小体都依赖于RIP1-RIP3-DRP1通路。如水疱性口炎病毒(VSV)、仙台病毒(Sendai Virus,SeV)、登革热病毒及poly(I:C)[31]。此外,线粒体抗病毒信号蛋白(MAVS)在病毒感染的情况下将肿瘤坏死因子受体作用因子3(TRAF3)招募到ASC,泛素化的ASC促进NLRP3寡聚,从而增强NLRP3炎症小体的激活[32]。NLRP3还显示在病毒感染期间与线粒体融合蛋白2直接相关[33]。然而,有报道认为,线粒体损伤引起的活性氧对于激活线粒体内的NLRP3不是必需的。相反,由EMCV感染诱导的线粒体膜电位是激活NLRP3 炎性小体所必需的。在适当的线粒体膜电位作用下,NLRP3转运至线粒体,与线粒体融合蛋白2结合,进而促使NLRP3炎性小体活化[34]。
病毒感染过程中,无论是溶酶体破坏还是线粒体损伤均诱导ROS产生。由此可见,ROS产生对NLRP3炎性小体激活起到了一定作用。已有研究证明了有些病毒依赖ROS的产生激活NLRP3炎性小体。如在HCV感染的细胞中加入ROS的清除剂,发现NLRP3炎性小体的激活受到抑制,提示ROS对NLRP3炎性小体的激活起正调节作用[35]。而对于ROS在NLRP3炎性小体激活中的具体作用仍需要探讨。
3.3 病毒孔蛋白介导的激活 除上述常见激活机制外,研究还发现有少数病毒能借助孔蛋白激活NLRP3炎性小体。病毒孔蛋白为病毒编码的离子通道蛋白,可与宿主细胞膜相互作用形成选择性离子通道,介导特殊离子(如Na+,K+,Ca2+,Cl-1)的运输,辅助病毒进入宿主细胞,进而激活炎性小体,促进炎症的发生。如甲型流感病毒(IAV)编码的质子特异性离子通道M2蛋白能促使酸化的高尔基体释放氢离子,导致高尔基体H+浓度失衡来启动NLRP3炎性小体的激活[36]。呼吸道合胞病毒(Respiratory syncytial virus,RSV)表达的SH蛋白能聚集在宿主细胞高尔基体的脂筏结构中,形成离子通道,进而诱导NLRP3从胞质移向高尔基体,触发炎性小体的激活[37]。脑心肌炎病毒编码的2B蛋白,定植于内质网和高尔基体,能使宿主细胞细胞器中的钙离子浓度下降,进而促使NLRP3炎性小体的组装和IL-1β的分泌。这一结论已在鼠骨髓来源的巨噬细胞中得到证实。但2B蛋白对NLRP3炎性小体激活过程的具体途径有待进一步探索[38]。同样,近年来已经被证实的严重急性呼吸综合征冠状病毒(Severe acute respiratory syndrome coronavirus,SARS-CoV),其亚基因组RNA编码的E蛋白能诱导细胞内离子浓度失衡以及内质网应激,进而导致NLRP3炎性小体的激活。研究显示,E蛋白能在内质网高尔基体中间腔或高尔基体膜中形成钙离子通道,并激活NLRP3炎性小体[39]。
综上所述,当病毒感染时,NLRP3炎性小体的激活机制,如图1所示。
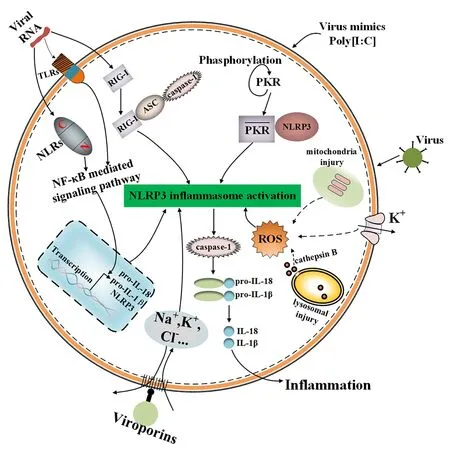
图1 病毒感染时NLRP3炎性小体激活路径模式图Fig.1 NLRP3 inflammasome activation pathway during viral infection
4 病毒感染负向调节NLRP3炎性小体活化
病毒不仅能通过编码相应的孔蛋白激活NLRP3炎性小体,然而研究发现,病毒编码的非结构蛋白能与炎性小体激活途径中的关键蛋白直接结合来抑制NLRP3炎性小体的激活。如麻疹病毒(Measles virus,MV)编码的V蛋白能与NLRP3蛋白直接结合来抑制NLRP3炎性小体的活化,其机制可能是通过阻碍NLRP3炎性复合体的组装过程进而抑制了炎性小体的活化。肠道病毒71型(Enterovirus type 71,EV-71)编码的2A和3C蛋白酶能对NLRP3蛋白进行切割以抑制NLRP3炎性小体的激活[41]。SARS-CoV-2基因组RNA翻译的非结构蛋白NSP1和NSP13,能显著抑制NLRP3炎性小体的活化。其机制可能是通过下调NLRP3、ASC、caspase-1和IL-1β等组分的表达来干扰NLRP3炎性小体的激活[42]。
病毒感染时,病毒不仅通过自身组分干扰NLRP3炎性小体的激活以逃避机体的免疫应答,近期有学者称,宿主的线粒体自噬也能抑制NLRP3炎性小体的激活。在自噬系统缺陷的小鼠血清中发现,IL-18、IL-1β含量明显升高,表明线粒体自噬对NLRP3炎性小体起负调控作用,因此,病毒感染自噬系统缺陷的小鼠可能更难以逃避机体的免疫应答机制[43]。
5 结语与展望
NLRP3炎性小体的激活对机体正常发挥免疫应答,防御病原体入侵至关重要。一方面,NLRP3炎性小体激活过程受阻,机体对外界病原体的侵袭不能及时或有效的产生应答,病原微生物不能被清除,从而导致机体感染。另一方面,NLRP3炎性小体过度激活,机体产生过量的炎性因子导致细胞过度焦亡以及严重的病理损伤。大部分的研究显示,相对NLRP3炎性小体的抑制机制而言,其激活更利于机体免疫功能的正常发挥,对宿主具有保护作用。实验用IAV感染NLRP3、ASC、caspase-1基因敲除的小鼠,发现小鼠的死亡率明显升高。提示NLRP3炎性小体在病毒感染机体时发挥了保护宿主的作用。
病毒感染与NLRP3炎性小体之间存在复杂的生物学关系。NLRP3分子作为一种病毒炎症信号(如病毒RNA或病毒离子孔道蛋白)的识别受体,在NLRP3炎性小体激活中发挥重要作用。 宿主细胞内NLRP3蛋白识别病毒炎症信号,诱导炎性复合体组装并触发其激活,促使下游炎性因子产生,抑制病毒在宿主细胞内繁殖,发挥抗病毒作用。同时,病毒也能依赖自身基因组编码的非结构蛋白,抑制NLRP3炎性小体的组装及活化,以逃避机体的免疫应答机制。虽然一些研究已经证实了某些病毒感染时,能刺激机体NLRP3炎性小体激活,然而我们对NLRP3炎性小体参与抗病毒免疫的复杂机制以及在激活过程中是否有其他分子参与还不完全了解。 因此,从分子水平深入研究病毒感染与NLRP3炎性小体之间的复杂机制,有利于我们从根本上控制病毒在宿主细胞中复制,为临床抗病毒感染、治疗及药物研发提供新的策略。
利益冲突:无
引用本文格式:曹旭,贾朝霞,刘浩,等. 病毒感染与NLRP3炎性小体[J].中国人兽共患病学报,2022,38(5):458-463. DOI:10.3969/j.issn.1002-2694.2022.00.056
