PM诱导支气管上皮细胞炎症反应及内质网应激机制研究
2022-10-12施强强董年刘莉王强陈俊杰陈成水
施强强 董年 刘莉 王强 陈俊杰 陈成水
作为空气污染物中的重要成分,细颗粒物(particulate matter,PM)的直径决定了其可以随呼吸气流进入人体肺脏,进而广泛沉积在气管-肺泡表面,诱导呼吸系统疾病的发生、发展[1]。气道上皮是呼吸系统抵御空气污染物的首要屏障,PM通过与气道上皮直接作用触发细胞毒性反应,导致气道上皮细胞过早产生炎症反应并凋亡坏死。气道上皮细胞在气道炎症的发生、发展中扮演了始动细胞和继发受体的双重角色[2-3]。目前关于PM诱导气道上皮细胞产生细胞毒性反应的病理生理机制尚未完全阐明。内质网应激(endoplasmic reticulum stress,ERS)以内质网稳态破坏、功能紊乱和错误折叠蛋白的堆积为特征,是近年来的研究热点。氧化应激、缺血再灌注等病理条件可能诱发细胞发生ERS。适度的ERS是细胞应对环境刺激的自我保护机制,但持续的ERS可能加重炎症反应,甚至导致细胞过早凋亡坏死[4-5]。本研究拟探讨在PM诱导下支气管上皮细胞的炎症反应及作用机制,为揭示气道炎症发病机制、探寻潜在诊治靶点提供参考。
1 材料和方法
1.1 材料6~8周SPF级雄性C57BL/6小鼠购自北京维通利华实验动物技术公司,许可证号为SXCK(京)2016-0011;人支气管上皮细胞BEAS-2B购自上海中科院细胞库。PM购自美国NIST公司,加入PBS中配置成4 g/L母液;RPMI 1640基础培养液、PBS、FBS购自美国Gibco公司;鼠抗人CCAAT增强子结合蛋白同源蛋白(CCAAT/enhancer binding protein,CHOP)抗体、兔抗人葡萄糖调节蛋白前体78(glucose-regulated protein 78,GRP78)抗体购自美国CST公司;IL-6、IL-8及前列腺素E2(prostaglandin E2,PGE2)ELISA检测试剂盒购自上海博蕴公司;活性氧(reactive oxygen species,ROS)检测试剂盒、脂质氧化丙二醛(malondialdehyde,MDA)检测试剂盒、N-乙酰半胱氨酸(N-acety-L-cysteine,NAC)购自上海碧云天生物技术有限公司;二喹啉甲酸(bicinchoninic acid,BCA)蛋白浓度检测试剂盒、预染蛋白Marker和ECL发光底物购自美国Thermo公司。
1.2 动物模型的建立及实验分组处理
1.2.1 动物分组和模型建立选取20只小鼠适应性喂养1周后,随机分成实验组和空白组,每组10只。0、24 h时空白组气道滴注PBS 25 μl,PM组根据动物体质量(按4 mg/kg剂量)气道滴注等体积PM悬混液。
1.2.2 检测标本的获取和处理末次气道滴注24 h后用4%的水合氯醛腹腔注射麻醉小鼠。取每组3只小鼠,经气管插管灌注0.9%氯化钠溶液,再缓慢回抽,重复3次,获取肺泡灌洗液,用以检测IL-6、IL-8和PGE2表达水平。另取每组3只小鼠,断颈处死后,打开胸腔获取新鲜肺组织,用以检测GRP78和CHOP表达水平。取每组4只小鼠,经气管插管灌注多聚甲醛,观察到小鼠双侧肺逐渐变膨隆,拉紧绳结封闭气管,获取肺组织进行HE染色和免疫组化分析。
1.3 BEAS-2B细胞分组和模型建立将BEAS-2B细胞培养于含有10%FBS和1%青霉素/链霉素的RPMI-1640培养基中,37℃,5%CO2,隔天换液,胰酶消化传代,获取生长状况良好的对数生长期细胞进行加药处理。细胞模型Ⅰ分为PM100组、PM200组和PM400组,分别加入由母液稀释的100、200和400 mg/L PM,并以不加PM为空白组。4组细胞培养1 h后检测ROS强度。细胞模型Ⅱ分为空白组、NAC组、PM组和PM+NAC组,NAC组和PM+NAC组加入2.5 mmol/L NAC,1 h后PM和PM+NAC组加入200 mg/L PM,空白组不加任何处理,4组细胞继续培养24 h后检测ROS强度和GRP78/CHOP表达水平。
1.4 小鼠观测指标
1.4.1 组织病理分级取两组小鼠肺组织常规切片,HE染色,光学显微镜下观察肺组织病理变化。以0~4分的主观评分评估支气管附近及血管附近的炎症等级[6]:正常组织,未见炎症细胞,为0分;偶见炎症细胞为1分;支气管或血管周围不均匀分布的炎症细胞或炎症细胞围绕成薄的环状(层厚1~5个炎症细胞),为2分;支气管或血管周围被炎症细胞围绕成厚的环状(层厚>5个炎症细胞),为3分;支气管、血管或肺泡分布弥漫聚集的炎症细胞,为4分。
1.4.2小鼠肺泡灌洗液中IL-6、IL-8和PGE2表达的检测采用ELISA法。收集两组小鼠肺泡灌洗液,2 000 r/min、4℃离心20 min后获取上清液,加注至ELISA板中。37℃孵育2 h后移去板孔中液体,添加生物素抗体,37℃继续孵育1 h;再次移去液体,添加HRP-抗生物素蛋白,37℃孵育1 h;重复洗涤5次,显色并终止。测定板孔液体450 nm波长下的吸光度,根据标准曲线计算样品IL-6、IL-8和PGE2的质量浓度。
1.4.3 小鼠肺组织中MDA表达的检测采用硫代巴比妥酸(thiobarbituric acid,TBA)法。MDA与TBA结合可形成红色的MDA-TBA,在530~540 nm处有最大吸收。根据MDA检测试剂盒方法,获取肺组织并剪切成细小碎片,加入适量裂解液充分裂解。添加0.2 ml由TBA配置的MDA检测工作液,100℃水浴15 min,冷却至室温后,测定532 nm处吸光度;采用BCA法检测该肺组织样品裂解液中的蛋白浓度,计算肺组织中MDA的质量摩尔浓度。
1.4.4 小鼠肺组织中GRP78和CHOP表达的检测采用Western blot法。收集各组小鼠肺组织,加入RIPA裂解液,于冰上研磨捣碎,14 000 r/min、4℃离心10 min后提取上清液。根据BCA蛋白浓度试剂盒测定总蛋白浓度,并将各组样品蛋白稀释至5 g/L,加入loading buffer后进行变性处理。变性好的蛋白样品按照每孔6 μl的上样量加入聚丙烯酰胺凝胶上样孔中,湿转至硝酸纤维素膜,在质量浓度为5%的脱脂牛奶中室温封闭2.0 h,一抗4℃孵育过夜,TBST缓冲液洗膜10 min,重复3次;二抗室温孵育1.5 h,再用TBST缓冲液洗膜10 min,重复3次。加入化学发光试剂,曝光显影。以β-肌动蛋白(β-actin)为内参,根据灰度值计算GRP78和CHOP的相对表达量。
1.4.5 小鼠肺组织中GRP78和CHOP表达的检测采用免疫组化法。获取肺组织石蜡切片,常规使用二甲苯脱蜡,双氧水处理去除内源性过氧化物酶,柠檬酸抗原组织抗原修复液处理切片修复抗原。常规血清封闭,一抗4℃孵育过夜,二抗室温孵育60 min,PBS清洗后使用二氨基联苯胺显色,以PBS代替一抗作为对照,中性树脂封片后正置显微镜下拍片,并用Image J软件进行分析,计算GRP78和CHOP的累计光密度值。
1.5 细胞检测指标
1.5.1 BEAS-2B细胞ROS水平检测根据ROS检测试剂盒方法进行。取细胞模型Ⅰ和Ⅱ各组细胞,与稀释1 000倍的ROS探针2,7-二氯荧光素二乙酸酯(DCFHDA)共培养20 min,使用高内涵成像分析系统(美国PerkinElmer公司)进行高内涵成像分析。DCFH-DA被细胞内的酯酶水解生成2',7'-二氯二氢荧光素(DCFH),细胞内的ROS可以氧化无荧光的DCFH生成有荧光的2',7'-二氯荧光素(DCF)。使用Image J软件检测DCF的荧光强度,判断细胞内ROS水平,绿色荧光越强则细胞氧化应激程度越高。
1.5.2 BEAS-2B细胞GRP78和CHOP表达的检测采用Western blot法。取细胞模型Ⅱ的4组细胞,裂解液裂解后,4℃、14 000 r/min离心10 min,取上清液。按照
1.4.4 方法测定并计算GRP78和CHOP的相对表达量。1.5.3 BEAS-2B细胞IL-6、IL-8和PGE2表达的检测采用ELISA法。取细胞模型Ⅱ的4组细胞,获取上清液后按照1.4.2方法测定并计算样品IL-6、IL-8和PGE2的质量浓度。
1.6 统计学处理使用SPSS 20.0统计软件。所有检测均重复3次。计量资料以表示,多组间比较采用单因素方差分析,组间两两比较采用Bonferroni校正的t检验。P<0.05为差异有统计学意义。
2 结果
2.1 两组小鼠支气管病理形态改变的比较PM组小鼠支气管周围炎症细胞浸润,支气管壁增厚,伴随PM气道沉积,见图1a。相较空白组,PM组病理分级评分升高,差异有统计学意义(P<0.01),见图1b。
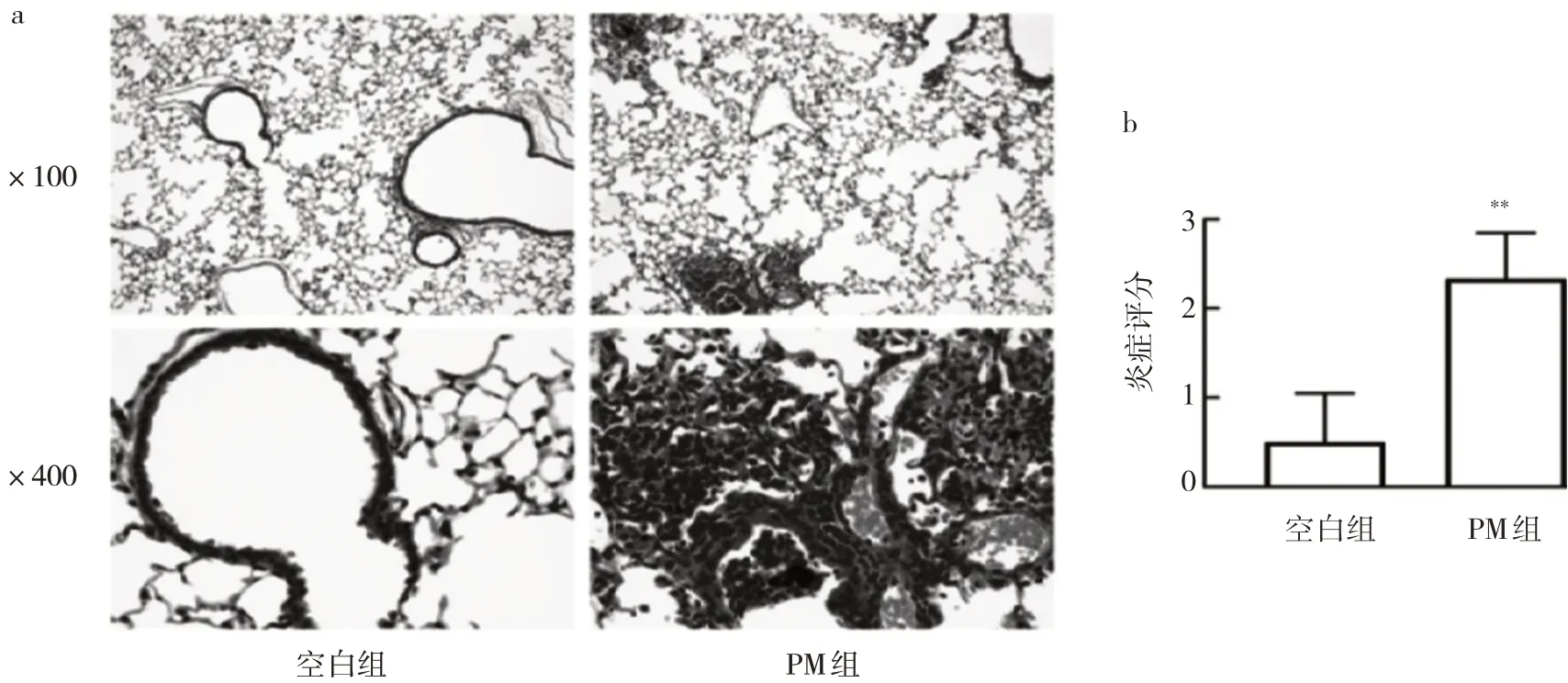
图1 两组小鼠支气管病理形态改变的比较(a:HE染色;b:炎症评分)
2.2 两组小鼠肺泡灌洗液中IL-6、IL-8和PGE2表达的比较相比空白组,PM组IL-6、IL-8和PGE2等炎症介质的质量浓度升高,差异均具有统计学意义(均P<0.01),见图2。

图2 两组小鼠肺泡灌洗液IL-6、IL-8和PGE2表达的比较
2.3 两组小鼠肺组织中MDA表达的比较相比空白组,PM组MDA质量摩尔浓度升高,差异有统计学意义(P<0.01),见图3。

图3 两组小鼠肺组织MDA表达的比较
2.4 两组小鼠肺组织CHOP和GRP78表达的比较Western blot检测结果可见,相比空白组,PM组肺组织CHOP和GRP78相对表达量升高,差异均有统计学意义(均P<0.05),见图4。

图4 两组小鼠肺组织GRP78/CHOP蛋白表达的比较(a:蛋白电泳图;b:GRP78相对表达量;c:CHOP相对表达量)
2.5 两组小鼠肺组织CHOP和GRP78表达的比较免疫组化染色结果可见,相比空白组,PM组棕黄色染色范围增加,染色程度增加,提示PM组细胞中CHOP和GRP78的表达升高,见图5a。相比空白组,PM组肺组织CHOP和GRP78累计光密度值升高,差异均有统计学意义(均P<0.05),见图5b-c。

图5 两组小鼠肺组织GRP78/CHOP蛋白表达的比较(a:肺组织免疫组化图,×400;b:CHOP的累计光密度值;c:GRP78的累计光密度值)
2.6 BEAS-2B各组细胞ROS水平的比较不同质量浓度PM作用下,BEAS-2B细胞内绿色荧光强度以剂量依赖的方式增加,见图6a;相比PM组,PM+NAC组细胞内绿色荧光强度减少,见图6b。与空白组相比,细胞模型Ⅰ的其他3组细胞ROS水平均升高,差异均有统计学意义(均P<0.01),见图6c。细胞模型Ⅱ中,与空白组相比,PM组细胞ROS水平升高,差异有统计学意义(P<0.01);与PM组相比,PM+NAC组细胞ROS水平降低,差异有统计学意义(P<0.05),见图6d。
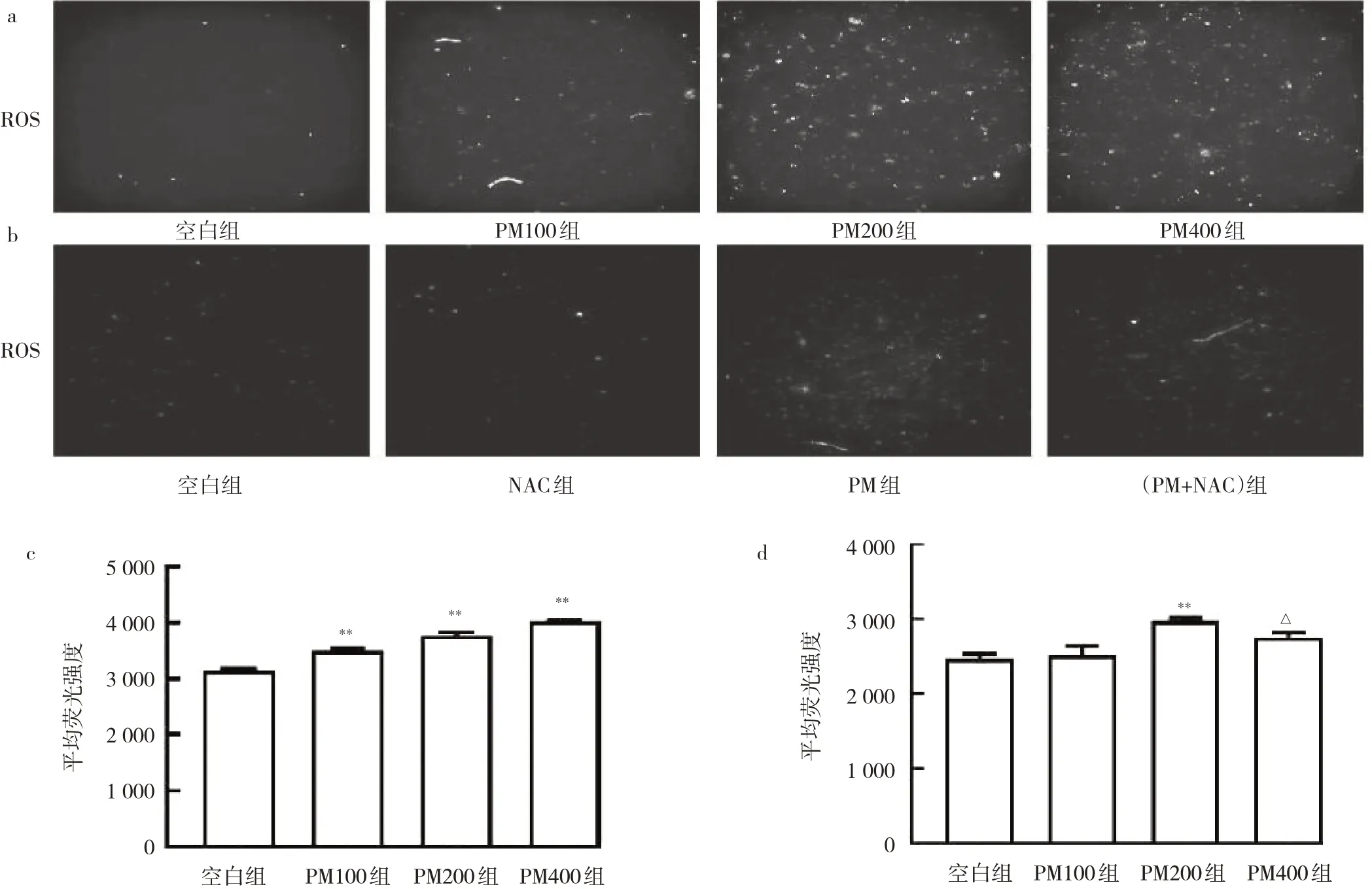
图6 4组BEAS-2B细胞ROS免疫荧光图像及其荧光强度
2.7 4组BEAS-2B细胞GRP78和CHOP表达的比较4组BEAS-2B细胞中均有GRP78和CHOP表达(图7a);相比空白组,PM组GRP78和CHOP相对表达量均升高,相比PM组,PM+NAC组GRP78和CHOP相对表达量均降低,差异均有统计学意义(P<0.01),见图7b-c。
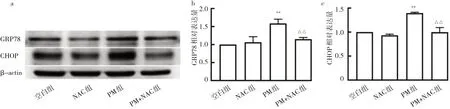
图7 4组BEAS-2B细胞GRP78和CHOP蛋白表达的比较(a:蛋白电泳图;b:GRP78相对表达量;c:CHOP相对表达量)
2.8 4组BEAS-2B细 胞IL-6、IL-8和PGE2表 达 的比较相比空白组,PM组IL-6、IL-8和PGE2质量浓度均升高,差异均有统计学意义(均P<0.01);相比PM组,PM+NAC组IL-6、IL-8和PGE2质量浓度均降低,差异均有统计学意义(均P<0.01),见图8。
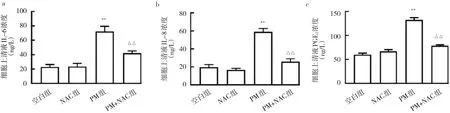
图8 4组BEAS-2B细胞上清液中IL-6、IL-8和PGE2表达的比较
3 讨论
中国成人肺部健康研究揭示PM是气道疾病独立的危险因素[7]。支气管上皮细胞是气道固有结构细胞,PM作用于支气管上皮细胞可触发系列细胞毒性反应,导致支气管上皮细胞产生氧化应激、细胞自噬和炎症损伤等功能状态的改变[8-10]。已有研究表明PM可诱导细胞产生氧化应激[11]。研究PM作用下支气管上皮细胞的病理生理改变,可为揭示气道疾病新的发病机制、寻找潜在的诊治靶点提供参考。
内质网是细胞中重要的细胞器之一,负责蛋白质的合成、折叠和翻译,在缺血低氧、钙离子紊乱、营养物质缺乏等病理状态下内质网功能可发生紊乱,导致未折叠和错误折叠蛋白质堆积,细胞在自我感知下会适应性启动内质网应激来恢复蛋白质平衡[12]。根据诱发原因,ERS可分为3类,一是由未折叠或错误折叠蛋白质在内质网腔内过度堆积引起的未折叠蛋白反应(unfold protein response,UPR),二是正确折叠的蛋白质在内质网腔内过度堆积引起NF-κB激活导致的内质网超负荷反应,三是因胆固醇缺乏引发固醇调节原件结合蛋白激活而产生的固醇调节级联反应[13]。UPR不仅可以通过减少蛋白质合成和促进错误折叠蛋白质降解,还可以上调参与蛋白质平衡调控的靶基因来缓解内质网压力[14]。在稳定状态下,UPR有助于降低内质网压力,维持细胞内稳态。然而ERS过强或持续时间过久,则最终加重炎症反应以及激活细胞死亡程序[4,15-16],因此内质网应激是细胞的自我保护性功能调整。GRP78被认为是ERS的主要标志性蛋白分子,当错误折叠蛋白积累时,GRP78从内质网压力传感器RNA依赖的蛋白激酶样内质网激酶[RNA-dependent protein kinase(PKR)-like ER kinase,PERK]、活化转录因子6(activating transcription factor 6,ATF6)和肌醇需求酶1(inositol requiring enzyme 1,IRE-l)中分离出来,激活ERS的3条信号通路[17]。因此,GRP78的表达升高被认为是ERS激活的标志;持续ERS导致CHOP表达,并诱导与B细胞淋巴瘤/白血病-2(B cell lymphoma/leukemia-2,BCL-2)蛋白家族促凋亡因子相互作用的细胞死亡调节蛋白(BCL-2 interacting mediator of cell death,BIM)的转录,BIM可通过增加线粒体膜通透性从而激活线粒体凋亡机制[18]。本研究从动物实验和细胞实验2个层面探讨了PM对ERS病理生理的诱导机制。PM气道滴注的动物模型和与PM共培养支气管上皮细胞BEA-2B中,均检测到了GRP78和CHOP表达升高,推测PM作用下支气管上皮细胞触发的ERS是持续性的,但升高表达的CHOP与PM诱导的支气管上皮细胞凋亡是否相关还未知。目前认为细胞在应激状态下发生ERS的分子机制涉及到氧化应激、丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)等信号通路[19-20]。氧化应激是指氧化自由基产生过多,打破了其与抗氧化剂之间的平衡。这种不平衡会导致重要的生物分子和细胞受损,最终导致细胞死亡[21-22]。最常见的2种氧化剂家族为ROS和活性氮(reactive nitrogen species,RNS)[23]。ROS为一系列分子氧的衍生物,其产生与新陈代谢和其他酶促过程有关,大多数ROS是通过线粒体呼吸链产生的,此外还依赖细黄嘌呤氧化酶系统、一氧化氮合成酶系统和烟酰胺腺嘌呤二核苷酸磷酸氧化酶系统(nicotinamide adenine dinucleotide phosphate oxidases,NOXs)[24-25]。适量的ROS可以促进细胞增殖、分化,并可作为第二信使激活植物和动物细胞中的信号通路以响应细胞内外环境条件的变化[26]。然而当ROS产生量超过机体调节能力时,则会引起炎症反应、氧化应激窘迫,从而引发一系列细胞生理功能紊乱,导致细胞损伤或死亡[27]。本研究利用高内涵成像分析系统发现,PM刺激下支气管上皮细胞ROS水平升高且呈现PM剂量依赖性。给予ROS抑制剂NAC干预后,ROS水平降低,可见NAC干预逆转了PM诱导的ERS,伴随着PM诱导的炎症介质的分泌改变。ROS的产生与慢性炎症相关,在炎症期间,肥大细胞和白细胞被募集到损伤部位,由于氧吸收量增加导致“呼吸爆发”,因此损伤部位ROS的释放和积累增加[28-29]。此外,在动脉粥样硬化、缺氧再灌注模型中,ROS作为第二信使能够促进炎症反应的发生,炎症介质产生增加[30]。但PM诱导的ERS与炎症介质分泌的相关性尚不清楚。
综上所述,本研究发现PM可以诱导气道炎症,伴随支气管上皮细胞产生ERS;PM诱导的氧化应激介导了ERS病理生理过程。后续研究可考虑使用雾化吸入的方法,以更好地模拟真实事件中PM暴露情况。ERS涉及复杂的蛋白调控网络,除CHOP和GRP78参与的未折叠蛋白反应外,ERS还包括肌醇需求酶-1(inosital-requiring enzyme 1)α途径、活化转录因子6(activating transcription factor 6,ATF6)途径和PERK途径,后续可进一步研究更精细的氧化应激蛋白和氧化应激后信号通路的改变,从而更加全面反映ERS病理状态。
