羟基氧化铟团簇与二氧化碳和甲烷作用的密度泛函理论研究
2022-08-22何鸿锐夏文生张庆红万惠霖
何鸿锐,夏文生,张庆红,万惠霖
(厦门大学化学化工学院,固体表面物理化学国家重点实验室,醇醚酯化工清洁生产国家工程实验室,福建省理论与计算化学重点实验室,厦门 361005)
经济发展和全球人口增长导致化石燃料消耗日增的同时,伴随着大量会引起全球气候环境恶化的温室气体(CH4,CO2等)的排放,因此,转化与利用CO2和作为化石能源中的气体燃料CH4分子的研究受到了极大关注[1~3]. 由于甲烷分子的化学稳定性,其目标产物较甲烷活泼而易进一步转化,故甲烷的转化与利用的主要问题是目标产物的选择性,如甲烷氧化偶联(OCM)制烃路线工业化发展的主要障碍,就在于催化反应(微放热)的动力学控制过程的改善[4,5];目前,CO2转化的研究主要集中于CO2加氢反应[6,7],而其中的关键是相对惰性的CO2分子的高效活化,如铜基催化剂催化CO2加氢制甲醇即具有选择性高和转化率极低的特点[8~12].
最近有研究指出,氧化铟基催化剂在CO2加氢反应中呈现出优异的催化性能,但对其作用机制尚不明确. 如Dang等[13]采用In2O3催化剂催化CO2加氢反应,产物甲醇选择性为92.4%,单程CO2转化率超过17%;Rui等[14]采用Au-In2O3催化CO2加氢反应,在498 K时甲醇选择性约为100%,甲醇产量高达0.47 gMeOH·h-1·,他们认为CO2能在较温和条件下得以有效活化的原因是,In2O3催化剂表面上氧空位的存在;然而,Posada-Borbon等[15]通过密度泛函理论(DFT)研究了H2和H2O存在下In2O3表面作用CO2的情况时,发现CO2吸附在无空位的羟基氧化铟表面上的活化能垒为46.31 kJ/mol,无羟基存在的In2O3表面上的活化能垒为161.13 kJ/mol(vs.144.73 kJ/mol,簇模型计算值[16]),即氧化铟上羟基的引入导致了CO2活化能垒降低. 同样,Ye等[17,18]采用DFT计算方法研究了H2存在下In2O3表面作用CO2时,也证实了In2O3表面发生了羟基化,并进而促进了CO2的吸附和转化. 甲烷C—H 活化在一些常规催化剂上的能垒通常较高,如Lei 等[19]采用氧化镧团簇模型进行DFT 计算,得到CH4的活化自由能垒为100.83 kJ/mol.
为了了解氧化铟基催化剂对CO2的作用机制,本文采用DFT方法,以含OH基团的氧化铟团簇为模型,通过计算研究了其与CO2作用过程的机制;为了了解CO2作为一种温和的氧化剂,其对CH4的C—H活化的影响. 对羟基氧化铟团簇与(CO2+CH4)的作用机制也进行了研究,以试图理解其涉及的C=O和C—H化学键的控制.
1 模型与计算方法
Wang 等[16]采用线型O—In—O、四元环In2O2和六元环In3O4等团簇模型,计算研究了其催化CO2加氢反应的过程,基于此,本文在这些团簇模型基础上构建了羟基氧化铟团簇,并采用Gaussian 09程序包[20]中DFT的B3LYP泛函[21,22]研究了羟基氧化铟团簇与CO2,CH4的作用. 其中,羟基氧化铟团簇中的In 原子采用SDD 基组[23],C,H 和O 原子采用Def2-TZVP 基组[24]. 相关的反应物、过渡态、产物和中间体的几何结构和自由能(含零点能校正)均在UB3LYP/SDD+Def2-TZVP水平下优化得到. 稳态和过渡态通过频率计算判断,采用内禀反应坐标(IRC)方法进行验证. 采用耦合簇理论(CCSD(T))[25~27]单点能计算提高所需能量的计算精度,相关的计算水平为UCCSD(T)//UB3LYP/SDD+Def2TZVP. 通过自然键轨道方法(NBO)[28]进行电荷分析.
以上述小团簇模型计算结果和In2O3(110)晶面结构为参照,构建包含50个原子(16个In原子、24个O原子和10个H原子)的扩展团簇模型. 考虑到计算效率,其中与CO2和CH4邻近并发生作用的In原子基组为SDD,C,H和O原子基组为Def2-SVP[29],其余未与底物分子邻近的In原子基组为Def2-SVP,O 原子基组为6-31G*[30]. 所用计算方法与小团簇情况相同. 相关的计算水平记为UB3LYP/(SDD+Def2SVP)∶UB3LYP/(Def2SVP+6-31G*).
所有能量和活化自由能垒均为298 K时吉布斯自由能(差)值. 积分网格采用Int=Ultrafine. 所有团簇的波函数均通过Stable=Opt予以检查并重新优化. 团簇的电子自旋多重度的选择则依据能量有利的原则确定(S:单重态;T:三重态).
2 结果与讨论
2.1 团簇的结构
Wagner等[31]通过实验和理论研究指出,氧化铟暴露于水汽中可以形成羟基氧化铟. 其DFT计算表明,氧化铟表面与水作用形成的羟基氧化铟可以稳定存在,而非接触探针原子力显微镜(AFM)的尖端探测结果则显示,羟基与In之间形成了较强的化学键.
图1给出了电中性团簇InO(OH),In2O2(OH)2,In2O3,In3O4(OH),In4O6和In4O5(OH)2等的几何构型和吉布斯自由能,这些团簇具有相对较低的能量(其它结构和相对能量见图S1,本文支持信息). 以单重态In2O2(OH)2(S)为例,其构型为双氧桥,而羟基处于末端的四元环,这是两个In原子以上的团簇所具有的共同特征[3个In以上的,还可能会出现三配位氧(3c)连接的结构];In—In键长(键级)为0.283 nm(0.104),即形成较弱的In—In,但其键长大于0.300 nm时,不会形成In—In键;In—OH键长(键级)为0.195 nm(0.767),说明In 与OH 形成了较强的化学键. 团簇中氧位点类型(相对于In 原子而言)可分为端末氧O(t)、同核单桥氧O(b1)、同核双桥氧O(b2)、同核三桥氧O(b3)和三配位氧O(3c).

Fig.1 Structures(singlet S, triplet T)/point group/Gibbs free energy(kJ/mol, 298 K) of (hydroxyl)indium oxide clusters optimized at the level of UB3LYP/SDD+Def2TZVPO(t), O(b1), O(b2), O(b3) and O(3c) stand for terminal, homonuclear single-bridged, homonuclear double-bridged, homonuclear triple-bridged and three-coordinated oxygen,respectively. Bond lengths(order)are in nm.
自旋多重度的改变引起(羟基)氧化铟团簇结构的变化不大[如图1的In2O2(OH)2(S/T)和In3O4(OH)(S/T):几何构型大体相同,O—In—O键角虽存在一些差异,但键长改变很小];在同等大小团簇中,低自旋团簇具有相对较低的吉布斯自由能.
羟基化前后团簇In—O 键长分别为0.203~0.226 和0.193~0.216 nm,无明显变化,与文献[15]报道一致;然而,羟基在团簇中的位置不同,所对应的团簇相对能量存在明显变化. H位于O(3c)上的团簇的能量相对较高,位于桥O(b)上的团簇次之,位于端末O(t)上的团簇最低(图S1).
2.2 (羟基)氧化铟团簇与CO2和CH4的作用
2.2.1 (羟基)氧化铟团簇与CO2的作用 (羟基)氧化铟团簇不同位点与CO2作用的过渡态结构如图2所示(产物结构见图S2,本文支持信息). 团簇In2O2(OH)2与CO2作用生成碳酸根的活性位点为In—O,过渡态具有四中心结构,其它团簇的情况类似. 这与文献[32]报道的无氧空位氧化铟与CO2作用的情况一致,CO2的C和O分别作用于氧化铟上的O和In,形成[2+2]四中心过渡态结构.
(羟基)氧化铟团簇的不同位点与CO2作用的能量学存在明显的差异(图2 和表1). 单重态团簇In2O2(OH)(S)与CO2作用的两种过渡态TS-CO2-In2O2(OH)2(S)(I)和TS-CO2-In2O2(OH)2(S)(II)的结构(图2),源于CO2分别作用于团簇的In—O(b2)和In—O(t)位点,而活化自由能垒(ΔGa)分别为16.99 和36.90 kJ/mol,反应自由能(ΔGr)分别为-31.63 和-17.20 kJ/mol,表明相对于端末羟基氧位点而言,团簇的桥氧位点与CO2的作用具有明显的能量学优势.

Fig.2 Optimized structures of the transition states for the interaction of singlet/triplet(S/T) clusters with CO2 at the level of UB3LYP/SDD+Def2TZVPBond lengths are in nm.
当作用位点不变,自旋多重度发生改变时,(羟基)氧化铟团簇与CO2作用的反应能量学也存在明显的变化(表1). 如InO(OH)(S)和InO(OH)(T)与CO2在位点In—O(t)上,活化自由能垒分别为8.87和39.58 kJ/mol,反应自由能分别为-98.62 和-54.18 kJ/mol;In2O2(OH)2(S)和In2O2(OH)2(T)与CO2在位点In—O(b2)上,活化自由能垒分别为16.99和41.51 kJ/mol,反应自由能分别为-31.63和6.36 kJ/mol,这些均表明低自旋团簇与CO2作用的活化自由能垒和反应自由能远低于相应的高自旋团簇,从而可有效促进反应的进行.
比较单重态InO(OH),In2O2(OH)2,In3O4(OH)和In4O5(OH)2等几个不同大小的团簇与CO2的作用,发现其最优途径的活化自由能垒分别为8.87,16.99,5.86和13.89 kJ/mol,对应的反应自由能分别为-98.62,-31.63,-66.32和-63.68 kJ/mol(表1). 即随着羟基氧化铟团簇的增大,其与CO2作用的活化自由能垒和反应自由能没有呈现明显的单向变化趋势,In为奇数的团簇与CO2作用的活化自由能垒似乎低于In为偶数的团簇,但反应自由能的变化却无相应的规律.

Table 1 Predicted activation free energy barrier(ΔGa) and reaction free energy(ΔGr) for the interaction of singlet/triplet(S/T)clusters with CO2 and CH4 at the level of UB3LYP/SDD+Def2TZVP and 298 K
为了说明羟基引入对CO2活化的影响,比较了羟基化前后的In2O3团簇与CO2作用的能量学. 如表1所示,CO2分别作用于团簇In2O2(OH)2(S)和In2O3(S)的In—O(b2)和In—O(b3)上所需的活化自由能垒为16.99 和87.11 kJ/mol,相应的反应自由能分别为-31.63 和-262.63 kJ/mol. 这表明羟基氧化铟团簇与CO2作用的活化自由能垒明显低于氧化铟团簇,即羟基的存在有利于氧化铟团簇对CO2的活化. 这与文献[15]结果相符.
2.2.2 (羟基)氧化铟团簇与CH4的作用 图3给出了(羟基)氧化铟团簇InO(OH),In2O2(OH)2,In2O3,In3O4(OH),In4O6和In4O5(OH)2与CH4作用的过渡态结构. CH4中的H与团簇中的O(b1),O(b2),O(b3)和O(3c)作用,CH3与In原子作用,形成[2+2]四中心结构过渡态. 与文献[33~35]报道类似.
(羟基)氧化铟团簇与CH4作用的能量学上的表现类似于CO2的情况(图3和表1),低自旋态的团簇的桥氧位点以及团簇中羟基的引入较有利于甲烷C—H的活化,而团簇的增大对甲烷C—H活化未呈现明显的单向变化趋势.

Fig.3 Optimized structures of the transition states for the interaction of singlet/triplet(S/T) clusters with CH4 at the level of UB3LYP/SDD+Def2TZVPBond lengths are in nm.
比较团簇对CO2和CH4两种小分子作用的情况,发现团簇作用于CO2的活化自由能垒低于CH4(表1). 如单/三重态团簇InO(OH)与CO2和CH4作用最优途径的活化自由能垒分别为8.87/39.58 kJ/mol和104.01/108.87 kJ/mol,反应自由能分别为-98.62/-54.18 kJ/mol和-197.57/15.77 kJ/mol;单/三重态团簇In2O2(OH)2与CO2和CH4作用的最低活化自由能垒分别为16.99/41.51 kJ/mol 和113.09/122.80 kJ/mol,反应自由能分别为-31.63/6.36 kJ/mol和-3.72/-74.52 kJ/mol. 均说明了团簇与CO2的作用要优于CH4,与Ma等[36]的研究观点一致.
2.2.3 (羟基)氧化铟团簇与CO2和CH4作用的电荷分析 表2 列出了CO2与(羟基)氧化铟团簇作用所涉及的一些原子或碎片上的电荷布居值. 由过渡态中团簇和CO2碎片上的电荷布居可见,CO2与(羟基)氧化铟团簇的作用,必伴随着电子由CO2流向氧化铟团簇或由羟基氧化铟团簇流向CO2. 因此,羟基的引入使氧化铟团簇活化CO2的作用机制由亲电加成改变为亲核加成.
通过比较表2中相同自旋态的团簇上不同位点与CO2作用,以及不同自旋态的团簇上相同位点与CO2作用涉及的电荷布居情况,发现团簇活性位点处的铟-δ+、氧-δ-的电荷差值(qIn-qO值)与CO2活化自由能垒的高低存在关联. 如自旋态相同的团簇In2O2(OH)2(S)活化CO2的两种位点为In—O(b2)和In—O(t),它们的qIn-qO值分别为3.443 和3.233,相应的CO2活化自由能垒分别为16.99 和36.90 kJ/mol;自旋态不同的团簇In2O2(OH)2(S)和In2O2(OH)2(T),活化CO2的活性位点In—O(b2)的qIn-qO值分别为3.443和3.182,相应的CO2活化自由能垒分别为16.99和41.51 kJ/mol,表明团簇上作用位点的电荷差值越大,其对CO2作用的活化自由能垒则越低,也说明了低自旋团簇上的桥氧位点对CO2的活化较有利.
同样的规律也适用于氧化铟团簇和羟基氧化铟团簇的比较,且不难发现羟基的引入会使得活性位点In—O中In所带正电荷增加,从而可增强活性位点In—O通过四中心[2+2]方式与CO2的作用. 如表2所示,单重态团簇In2O3和In2O2(OH)2作用于CO2的最优途径的位点原子In/O 上的电荷qIn/qO分别为1.878/-1.131和2.077/-1.366,相应的活化自由能垒分别为87.11和16.99 kJ/mol,即羟基引入伴随的团簇活性位点间的电荷差值增大,导致了其对CO2作用的活化自由能垒的降低.
CH4与(羟基)氧化铟团簇作用的电荷分析情况与CO2类似(表3),即羟基的引入改变了团簇活化CH4的作用机制,氧化铟团簇活化CH4的方式为亲电加成,而羟基氧化铟团簇活化CH4的方式为亲核加成;团簇中铟-δ+、氧-δ-的电荷差值(qIn-qO值)越大,甲烷C—H的活化自由能垒越低,因此,低自旋的羟基氧化铟团簇上的桥氧位点对CH4的活化较有利.

Table 2 Natural bond orbital(NBO) charge population(q) and transition state(TS) frequency(vTS) for the interaction of CO2 with singlet/triplet(S/T)indium oxyhydroxide clusters and the activation free energy barrier(ΔGa,298 K)at the level of UB3LYP/SDD+Def2TZVP
2.2.4 (羟基)氧化铟团簇与(CO2+CH4)的作用 团簇活化(CO2+CH4)的可能方式是先活化CO2、后活化CH4,或先活化CH4、后活化CO2,基于上述的团簇分别活化CO2和CH4的结果,活化CO2具有明显的能量学上的相对优势,下面仅考虑团簇优先活化CO2的情况.
为了便于考察CO2的存在对CH4在团簇上活化的影响,考虑了团簇活化CO2后,CH4在团簇上邻近CO2处的几种可能落位,图S3(见本文支持信息)给出了甲烷C—H在团簇上活化的过渡态结构(活化自由能垒和反应自由能见表S1,本文支持信息). 可见,CO2存在下CH4作用于单/三重态团簇In3O4(OH)的位点为In—O(b2)和In—O(3c),具有较低的活化自由能垒和反应自由能的位点为铟-桥氧In—O(b2);而甲烷在其它团簇上则仅存一种落位,即铟-桥氧位点,与无CO2存在的情况相似. 团簇与CO2作用后,继续活化CH4的模式仍为[2+2],过渡态为团簇的位点In—O与甲烷H3C—H作用形成的四中心结构.
CO2存在下的单/三重态团簇InO(OH)和In2O2(OH)2与CH4作用的最优途径活化自由能垒分别为120.00/122.93和107.82/109.33 kJ/mol(表S1);无CO2存在下的单/三重态团簇InO(OH)和In2O2(OH)2与CH4作用的最优途径活化自由能垒分别为104.01/108.87 kJ/mol和113.09/122.80 kJ/mol,显示出CO2的存在与否对团簇作用于CH4的活化自由能垒影响不大,与Hoch等[37]的研究相符.

Table 3 Natural bond orbital(NBO) charge population(q) and transition state(TS) frequency(vTS) for the interaction of CH4 with singlet/triplet(S/T) indium oxyhydroxide clusters and the activation free energy barrier(ΔGa,298 K)at the level of UB3LYP/SDD+Def2TZVP
图4给出了(羟基)氧化铟与(CO2+CH4)作用的最优途径吉布斯自由能的变化. 可见,这个作用由CO2与团簇的In—O 作用(Step 1)、CH4与团簇上其它In—O 位点的作用(Step 2)和CH3从团簇上脱离(Step 3)3步构成,其中甲烷C—H活化和CH3脱离为关键步骤;采用CCSD(T)和B3LYP两种水平计算的各步骤的吉布斯自由能相对值G1-CCSD(T)和G2-B3LYP基本一致.
比较图4中不同自旋态团簇与(CO2+CH4)的作用,低自旋团簇具有明显的能量学优势. 单/三重态团簇InO(OH)(S/T)与CO2和CH4先后作用的活化自由能垒ΔG1a分别为3.69/45.23 kJ/mol 和111.93/121.04 kJ/mol,反应自由能ΔG1r分别为-85.60/-48.24 kJ/mol和-72.67/-58.11kJ/mol,显示出单重态羟基氧化铟团簇与(CO2+CH4)的作用在能量学上优于三重态,即低自旋团簇活化(CO2+CH4)较有利.
羟基的引入会促进团簇与(CO2+CH4)的作用. 如团簇In2O3(S)/In2O2(OH)2(S)与CO2和CH4的活化自由能垒ΔG1a分别为82.26/12.64 kJ/mol 和128.03/114.19 kJ/mol,反应自由能ΔG1r分别为-247.94/-18.20 kJ/mol 和-273.26/-77.74 kJ/mol,说明了羟基的参与降低了团簇与CO2和CH4依次作用的活化自由能垒,反应自由能值也有所降低,即羟基的存在使团簇与(CO2+CH4)的作用得到明显的加强,促进了二氧化碳和甲烷分子的活化.
2.2.5 羟基氧化铟扩展团簇与(CO2+CH4)的作用 小团簇大小的改变未对活化CO2和CH4的难易呈现出明显的单向变化趋势,而为了考察活化(CO2+CH4)分子的团簇的边界效应,构建了两个通过截取In2O3(110)晶面结构的部分而形成的扩展团簇(图5),其中球棍模型所示的团簇碎片InO(OH)和In2O2(OH)2邻近(CO2+CH4)分子;图6 所示为扩展团簇与(CO2+CH4)作用的能量学. 可见,InO(OH)和In2O2(OH)2相关的扩展团簇与CO2作用的活化自由能垒分别为53.43和79.04 kJ/mol,反应自由能垒分别为10.88和11.55 kJ/mol;与CH4作用的活化自由能垒分别为149.95和145.69 kJ/mol,反应自由能分别为29.92 和82.22 kJ/mol,遵循与小团簇相同的变化趋势. 即羟基氧化铟团簇与CO2作用的能量较低,而与CH4作用的能量相对偏高. 最近,Ma等[36]采用平板模型和密度泛函理论计算方法,获得在CO2存在下甲烷C—H的活化自由能垒为142.93 kJ/mol,与本文的计算值(145.69 kJ/mol)非常接近.

Fig.4 Predicted relative Gibbs free energy G1 and G2(kJ/mol, 298 K) for the interaction of singlet/triplet(S/T) clusters with (CO2+CH4) at the level of UCCSD(T)//UB3LYP/SDD+Def2TZVP and UB3LYP/SDD+Def2TZVP,respectively

Fig.5 Extended cluster model derived to InO(OH)(A)and In2O2(OH)2(B)
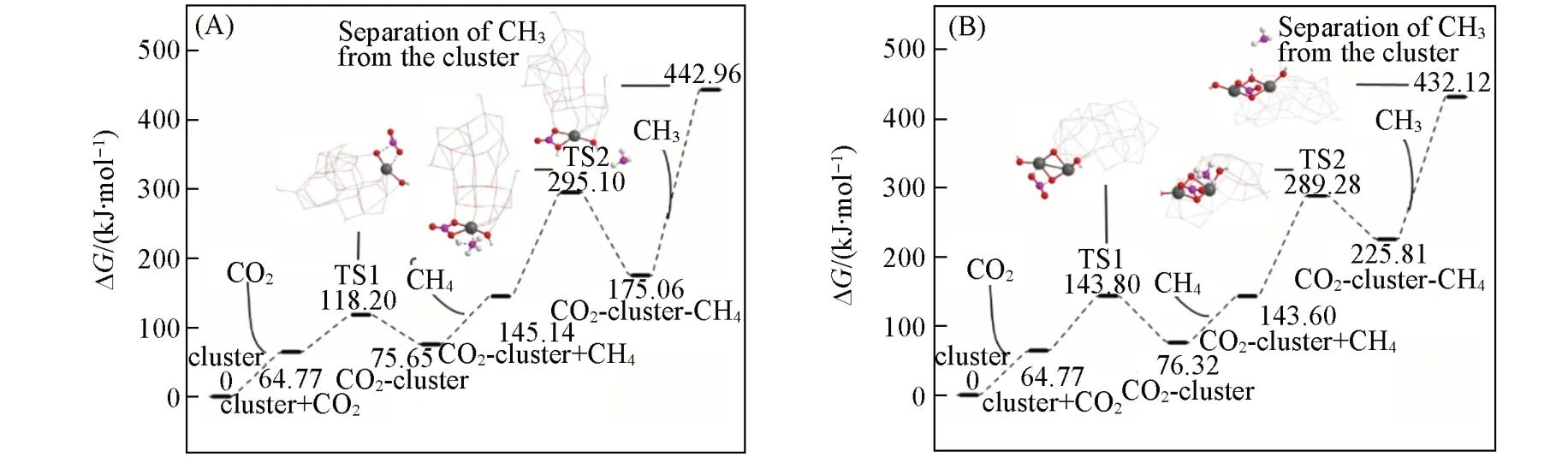
Fig.6 Predicted Gibbs free energy(ΔG, 298 K) for the interaction of extended clusters derived to InO(OH)(A) and In2O2(OH)2(B) with (CO2+CH4) at the level of UB3LYP/(SDD+Def2SVP) :UB3LYP/(Def2SVP+6⁃31G*)
3 结论
以(羟基)氧化铟团簇为模型,采用DFT方法,对氧化铟与CO2和CH4作用的机制进行了计算研究.结果表明,氧化铟活化CO2和CH4分子的最优活性位点为—In—O(桥氧)—,活化模式为[2+2]加成,过渡态具有四中心结构;羟基的存在可以显著降低氧化铟活化CO2和CH4分子的活化自由能垒,从而促进CO2和CH4分子中C=O和C—H的活化. 其根本原因是,羟基的引入可有效调节氧化铟活性位点上的电荷,使得氧化铟与CO2和CH4分子作用中涉及的电子流向发生了改变,羟基引入前,电子由CO2和CH4流向氧化铟(亲电活化),而羟基引入后,电子则由氧化铟流向CO2和CH4(亲核活化). 进一步的分析揭示,氧化铟活性位点—In—O(桥氧)—中的In和O间的电荷差值越大,其活化CO2和CH4分子的活化自由能垒越低,越有利于其对CO2和CH4分子的活化;具有较低电子自旋态的氧化铟活化CO2和CH4的能量学优势较明显. 氧化铟活化CO2明显优于CH4,而CO2的存在对其活化CH4的难易不会产生大的影响.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20220196.
