FLNA基因p.A267T变异首发与中国汉族人群耳-腭-指综合征相关
2022-08-18王静郑好胡清强张玉婷孔旭辉汪羽储九圣庞秀红
王静郑好胡清强张玉婷孔旭辉汪羽储九圣庞秀红*
1南京医科大学附属泰州市人民医院耳鼻咽喉-头颈外科(江苏 225300)
2大连医科大学(辽宁 116027)
3南通大学医学院(江苏 226019)
耳-腭-指综合征谱系疾病(Oto-palato-digital syndrome spectrum disorders,OPDSD)是一类罕见X连锁显性遗传性综合征型耳聋[1],其特征性表型主要涉及耳聋、颅面畸形及指趾畸形等多个方面,具体包括:耳聋、腭裂、前额突出、眼睑下垂、指趾畸形、胸廓畸形等[2]。目前分为以下五个综合征:耳-腭-指(趾)综合征1型(OPD1,OMIM#311300)、耳-腭-指(趾)综合征2型(OPD2,OMIM#304120)、Melnick-Needles综合征(MNS,OMIM#309350)、FMD1(OMIM#305620)和DCD(OMIM#300244)。其中,OPD1和OPD2合称为耳-腭-指综合征(Otopalato-digital syndrome,OPDS)[2,3]。男性患者表型严重程度与亚型有关:OPD2和MNS男性患者表型较重,在胚胎时期死亡或出生不久死亡;而OPD1和FMD1的男性患者可以存活且临床表型较轻微。对于女性,由于具有不同程度X染色体失活,患者表型特征差异性很大。因此,如果一个家系内没有男性患者,很难区分OPD1和OPD2[4-6]。另外,OPDS基因型和表型异质性都很高,同一基因位点突变会有不同的表型,而同一表型也有可能是由不同突变位点所导致。
FLNA(#OMIM,300017),位于 X染色体,包含48个外显子,编码细丝蛋白A(Filamin A),分子量为280KD,长度为160nm,含有2647个氨基酸[7]。Filamin A是一种肌动蛋白结合蛋白,由两个亚基自行连接形成二聚体,整个蛋白分子呈现“V”字形,每个亚基都含有N端的肌动蛋白结合域(ABD)和24个重复β-折叠片层结构。这24个重复片层结构被两个钙蛋白酶敏感性区域分成三部分:杆1区域、杆2区域和自行连接区[8],而N端的肌动蛋白结合域(ABD)则由CH1和CH2组成[9]。亚基上的肌动蛋白结合位点和伴侣蛋白结合位点参与调控细胞信号转导、维持细胞骨架等多种细胞活动[10]。Filamin A结构复杂性决定OPDS表型多样性[5]。目前所报道致病突变约340个,其中38个与OPD1相关(https://www.ncbi.nlm.nih.gov/clinvar)。
本课题组前期收集到一例中国汉族人群疑似OPDS家系成员样本,该家系三代内共有7名患者具有OPDS典型表型。本研究拟利用目前常用测序技术对该家系遗传致病性因素进行探寻,从而为遗传咨询和婚育指导提供理论依据,实现OPDS综合征型耳聋的一级预防。
1 材料与方法
1.1 伦理声明
本研究涉及到的先证者及家系成员均收集于泰州市人民医院耳鼻咽喉-头颈外科,所有患者或法定监护人均同意参与本研究并签署书面知情同意书。本研究得到了泰州市人民医院伦理委员会的批准,并符合“赫尔辛基宣言”。
1.2 病史询问及体格检查
本课题组前期所收集中国汉族疑似OPDS综合征家系的三代成员中均有患者。先证者为16岁女性,因“双耳听力下降、特殊面容、指趾畸形”就诊于泰州市人民医院耳鼻咽喉-头颈外科门诊。经详细病史询问,发现该先证者外祖母(已离世)、母亲及母亲其他四个姐妹均存在类似临床表型。对自愿参与研究的患病先证者III-1、母亲II-2、姨妈II-3以及正常表型父亲II-1和弟弟III-2进行全面体格检查,特别注意颅面部、耳科、眼科、骨骼系统等方面表型特征。利用颞骨高分辨电子计算机断层扫描(HRCT)以检查内耳及中耳可能异常。利用全身数字化X射线摄影进行骨骼检查;利用纯音测听、声导抗等进行听力学检查(图1)。
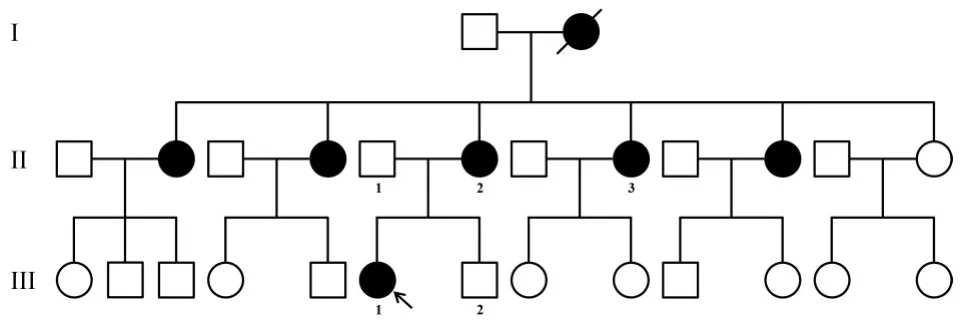
图1 家系图。正方形:男性;圆形:女性;白色图形:正常听力家系成员;黑色图形:患者;黑色箭头:先证者。Fig.1 Pedigree of a family.Square:male;Circle:female;White figure:normal hearing family members;Black figure:patients;Black arrow:proband.
1.3 采集外周静脉血及抽提DNA
EDTA抗凝管采集自愿参与研究的家系成员III-1、III-2、II-1、II-2和II-3外周静脉血各4ml。血液基因组DNA提取试剂盒(DP318-03,北京天根生化科技有限公司)抽提DNA。NanoDrop2000测定DNA浓度,-20℃保存备用[11]。
1.4 三大常见耳聋基因全序列筛查
利用先证者样本对中国汉族人群三大常见耳聋基因GJB2、SLC26A4及MT-RNR1所有编码外显子及其侧翼序列进行筛查,利用Sanger测序技术对凝胶电泳鉴别成功的PCR原液进行一代测序,利用Sequencher5.4软件进行结果判读[12,13]。
1.5 所有已知耳聋基因外显子靶向捕获二代测序
对常见耳聋基因筛查未发现致病突变的先证者样本,利用外显子靶向捕获二代测序技术对406个已知耳聋基因进行筛查。
选取3μg外周血DNA样本,打断为150 bp小样本,经补平、纯化、添加A碱基、连接adaptor、PCR扩增等步骤得到最终DNA文库。将DNA文库与相关液相捕获试剂盒混合,经吸附、洗脱等步骤得到目的基因片段。对406个已知耳聋基因外显子及其侧翼序列进行高通量测序[14]。
去除原始短序列中的接头和低质量数据后利用工具软件对可疑突变进行相关注释,最终得到候选致病突变[14]。
1.6 家系内验证及致病性分析
利用Sanger测序对可疑错义突变进行家系成员内验证,对基因型-表型共分离位点进行多物种保守性分析,并利用 SIFT、Polyphen2、Mutation Taster、PROVEAN等蛋白质功能预测软件进行致病性预测。
2 结果
2.1 临床表型
本研究家系所有患者均具有耳聋、特殊面容及指趾畸形等OPDS典型表型。先证者III-1自出生便具有OPDS特殊面容,包括前额突出、眼距宽大、眼睑下垂等(图2);2岁时发现双侧传导性听力下降(图3),否认耳毒性药物使用史及急慢性中耳炎等导致听力下降疾病史;另外,先证者具有鸡胸、“带状”肋骨、杵状指、跖趾畸形等骨骼畸形(图2)。有趣的是,先证者还具有牙齿早脱这一罕见表型。先证者母亲II-2和姨妈II-3均有先证者特殊面部表型及不同程度混合性听力下降。先证者父亲(II-1)和弟弟(III-2)未发现异常表型。

图2 先证者表型。a)胸部DR:白色箭头:“带状”肋骨畸形;b)特殊面容:白色实线箭头:前额突出;白色虚线箭头:眼睑下垂;黑色箭头:小颌畸形。c)和f)手指表型及影像学表现:白色箭头:杵状指;黑色箭头:手指弯曲畸形。d)和g)跖趾表型及影像学表现:跖趾弯曲畸形。e)颞骨HRCT:白色箭头:听小骨畸形。Fig.2 Phenotype of proband.a)DR in Chest:The white arrow indicates“ribbon”ribs;b)Special facial features:The white solid arrow represents prominent forehead;White dotted arrow represents drooping eyelid;Black arrow represents micrognathia;c)and f)Phenotypes and Radiologic images of fingers:The white arrows indicate Clubbing fingers and black arrows indicate finger bending;d)and g)Phenotypes and Radiologic images of feet and toes:Toe bending deformity;e)HRCT of temporal bone:The white arrow represents auditory ossicular deformity.

图3 纯音测听结果。BC:骨导;AC:气导。Fig.3 Pure tone audiometry.BC:bone conduction;AC:air conduction.
2.2 三大常见耳聋基因全序列筛查
对先证者DNA样本进行三大常见耳聋基因GJB2、SLC26A4及MT-RNR1全序列筛查,未发现任何致病突变。
2.3 已知耳聋基因二代检测
先证者DNA样本406个已知耳聋基因外显子捕获二代测序发现FLNA基因5号外显子杂合突变c.799G>A(p.A267T)。Sanger测序家系内验证发现该突变在所有自愿参与本研究的家系成员中呈现基因型-表型共分离,及患病先证者(III-1)、母亲(II-2)及姨妈(II-3)携带FLNA基因p.A267T突变,而表型正常的父亲(II-1)和弟弟(III-2)则未发现携带(图4)。

图4 突变峰图。Fig.4 Chromatograms of the core family.
2.4 致病性分析
FLNA基因c.799G>A(p.A267T)突变为编码区第799个核苷酸鸟嘌呤变异为腺嘌呤,导致第267位点的丙氨酸变异为苏氨酸。
1000Genomes、gnomAD和Exome Variant Server数据库无该位点信息。SIFT、PROVEAN、Mutation Taster及PolyPhen-2等软件预测结果显示致病可能性大(表1)。保守性分析显示位点p.A267在人、小鼠、大鼠、牛、猕猴及非洲蟾蜍物种中呈现高度保守(图5)。

图5 FLNA基因多物种保守性分析显示位点p.A267在多物种中高度保守。Fig.5 Multi-species conservative analysis of FLNA showing the highly conserved A267 residue(pointed by the arrow).
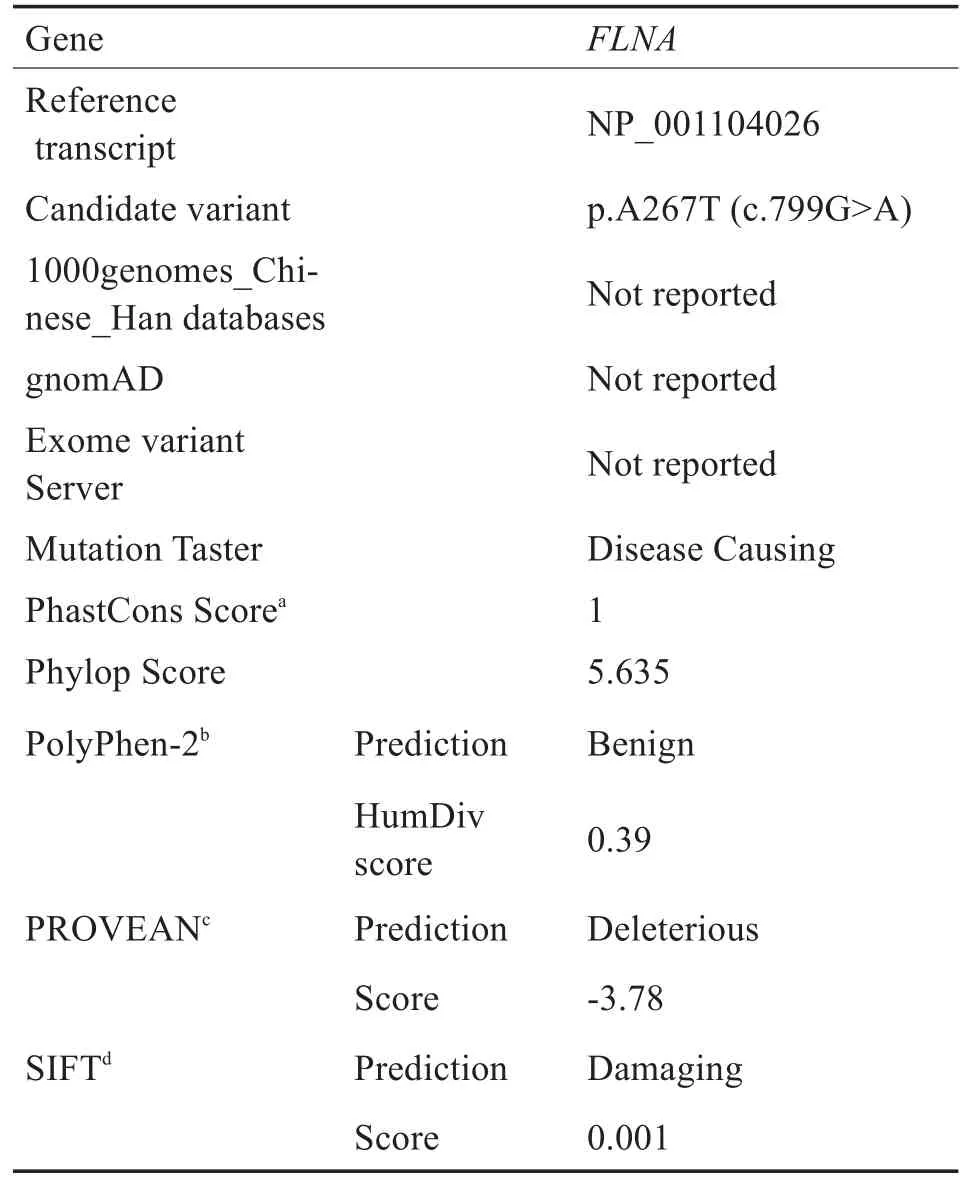
表1 该家系FLNA基因的致病性变异分析结果Table 1 Computational analysis of the p.A267T mutation in FLNA
3 讨论
OPDS是一类以不同程度骨-软骨发育不良为特征同时伴有骨外表现的一类疾病,其典型表型包括耳聋、面部畸形和指趾畸形等100余种[1]。本研究先证者具有双侧传导性耳聋、前额突出、眼距宽大、眼睑下垂和指趾畸形等表型,与既往文献报道OPDS患者特征性表型高度吻合[2]。另外,患者还具有“牙齿早脱”表型,既往文献报道OPDS患者具有“牙列不齐、缺齿”等牙齿发育不良的表型[15,16],但并未有过“牙齿早脱”的报道,推测其也属于OPDS牙齿发育不良表型之一。而先证者母亲及姨妈并无“牙齿早脱”,提示本研究家系内表型具有异质性,再次证实既往研究成果[1]。OPD2亚型男性患者具有幼年致死性,患者往往胚胎时期死亡或出生后不久死亡。而OPD1亚型男性患者表型较轻微且可以成活。在女性患者中,由于FLNA基因具有不同程度X染色体失活,表型更加复杂多变。两种亚型主要鉴别依据为患病家系中是否具有男性患者,如有男性患病成员能成活且表型较轻微则该家系为OPD1可能性较大;如有男性患病成员但胚胎时期或者幼年死亡且表型较严重,则该家系为OPD2可能性较大,如无男性患病成员则很难确定疾病亚型[17]。本研究家系内无男性患者,但鉴于该家系内所有患病女性成员表型轻微,且正常生活存活至今,因此推测该家系为OPD1的可能性较大。
迄今为止,FLNA基因p.A267T变异仅在欧洲人群的OPDS家系中报道一次,推测该位点可能与OPDS相关,未进行致病性分析及致病性基础研究[18]。发现该变异在多物种中位于绝对保守区域(图 5),SIFT、PROVEAN、Mutation Taster及 Poly-Phen2等工具软件预测其致病可能性大(表1)。本研究首次在中国汉族人群中发现该位点变异并提出其很可能为OPDS的致病突变,理由如下:1)在表型高度疑似OPDS家系中发现与OPDS相关的致病基因FLNA错义变异;2)该错义变异在家系内呈现基因型-表型共分离;3)既往报道中该变异亦与OPDS相关;4)工具软件致病性预测提示致病可能性大。本研究进一步为FLNA基因p.A267T变异导致OPDS提供了依据,该变异很可能为本研究家系遗传性病因。
前期研究发现导致OPDSD的突变类型一般为点突变和小片段缺失。这些突变并不影响Filamin A的丰度,而是导致蛋白介导某些功能的改变。另外,FLNA基因突变位点具有明显区域差异,如OPD1突变位点集中在外显子3、4或5;OPD2突变位点集中于外显子4、5或29等[17]。变异p.A267T位于5号外显子,编码CH2区域附近。发生在CH2区域的突变会导致Filamin A磷酸化增加及干扰肌动蛋白与Filamin A相互作用,从而导致肌动蛋白细胞骨架不稳定和信号转导中断[17]。既往,CH2区域的c.760G>A变异破坏ABD结构域中的负性调控元件,从而增强Filamin A对肌动蛋白的结合作用,即功能增益效应[17,19]。本研究所发现的FLNA基因p.A267T正位于CH2区域附近,并且很接近既往报道的c.760G>A突变位点,因此推测其致病机制很可能也与功能增益效应相关。为明确该突变位点深在致病机制,本课题组拟构建FLNAc.799G>A突变敲入小鼠模型进行在体功能研究。接下来,本研究将对该突变致病机制进行基础性研究,从而进一步明确具体致病机制。
OPDS遗传方式为X连锁显性遗传,其后代患病率为50%,以此为依据对本研究家系进行遗传咨询和婚育指导。本研究所发现FLNAc.799G>A杂合突变很可能为先证者及其家系患病成员遗传性病因。根据X连锁显性遗传规律,家系患病成员后代50%机率患病,后代患病风险高。建议利用胚胎植入前诊断、无创或有创产前诊断等技术避免后代耳聋,从而实现OPDS综合征耳聋的一级预防[20,21]。
4 结论
FLNA基因c.799G>A(p.A267T)变异很可能为本研究高度疑似OPDS家系的遗传性致病性因素。氨基酸改变所致功能增益效应很可能为该变异潜在致病机制,其深在致病机制将进一步利用FLNAc.799G>A突变敲入小鼠模型进行探索。根据家系患病成员表型严重程度和变异位点所在区域,该家系很有可能为OPD1。根据X连锁显性遗传性特点对该家系进行遗传咨询和婚育指导,从而实现OPDS综合征性耳聋的一级预防。
