理论计算在聚对苯二甲酸乙二醇酯(PET)塑料生物酶解聚中的应用进展
2022-08-13郑明娜董维亮李延伟张庆竹
郑明娜,董维亮,李延伟,张庆竹
(1.山东大学 环境研究院,山东 青岛 266237;2.南京工业大学 生物与制药工程学院,江苏 南京 211800)
塑料是一种重要的有机高分子聚合物,具有质量轻、耐腐蚀和价格低等优良特性,已被广泛应用于日常生活的方方面面[1-3]。自20世纪以来,全球塑料产量急剧增加,仅2020年产量就达到了3.67亿t[4],环境中大量的塑料废弃物对全球生态系统产生了巨大威胁[2,5-7]。当前塑料废弃物主要通过填埋、焚烧、机械或化学回收等方法进行处理。填埋和焚烧的方法虽然可以快速处理混合废旧塑料,但难以从根本上解决塑料污染问题[8]。机械回收具有操作简单、投资少及对环境影响小等优点,然而同时也存在回收种类有限、降级利用等问题[9-12]。化学回收比机械回收应用范围更广,但存在能耗高、二次污染等问题[11,13-15]。相比之下,塑料的生物解聚具有能耗低、绿色环保等优点,逐渐受到人们的青睐。生物解聚技术的关键是在微生物或酶作用下的各类塑料解聚反应[8,11,16-17]。
聚对苯二甲酸乙二醇酯(PET)是年生产量最大、应用最广泛的一类聚酯塑料。废弃PET塑料约占全球固体废弃物的12%,其生物解聚再利用是当前研究的热点[18-31]。自2005年首次报道PET的酶解聚成果以来,目前已经证实TfH、TfCut2、LCC、Cut190、HiC和IsPETase等多种酶均具有解聚PET的能力[22-23,32-38]。然而,酶的催化效率低、热稳定性差和经济可行性等因素在一定程度上制约了PET生物解聚技术的推广应用[20,31,39]。近年来,科研工作者利用X线衍射等技术解析了TfCut2[40]、LCC[22,41-42]、IsPETase[24,43]、BurPL[23]、PET2[44]和RgPETase[45]等多个PET解聚酶的三维结构,为深入理解PET的解聚机制并理性指导新酶的设计提供了重要的结构基础。
在实验研究方法迅速发展的同时,理论计算方法也取得了较快的发展。理论计算的优势是能够在分子层面研究解聚酶的动态性质及解聚机制,并预测潜在的有益突变位点。当前PET的酶催化解聚领域应用较多的理论计算技术主要包括分子对接技术(molecular docking)、分子动力学模拟技术(molecular dynamics,MD)、量子化学计算技术(quantum chemistry)及多尺度模拟技术(multiscale modelling)等[22,43,46-54],如Tournier等[22]采用分子动力学模拟技术阐释了极具工业化前景的LCCICCG突变体高效解聚PET的原因,Li等[55]将分子动力学模拟与机器学习模型相结合显著提升了TfCut2的解聚效率。图1为笔者以“plastic & biodegrad* & enzyme*”为主题词在Web of Science核心集搜索总结的近十年具有代表性PET解聚酶的鉴别、晶体解析以及工程化的文章出版情况。
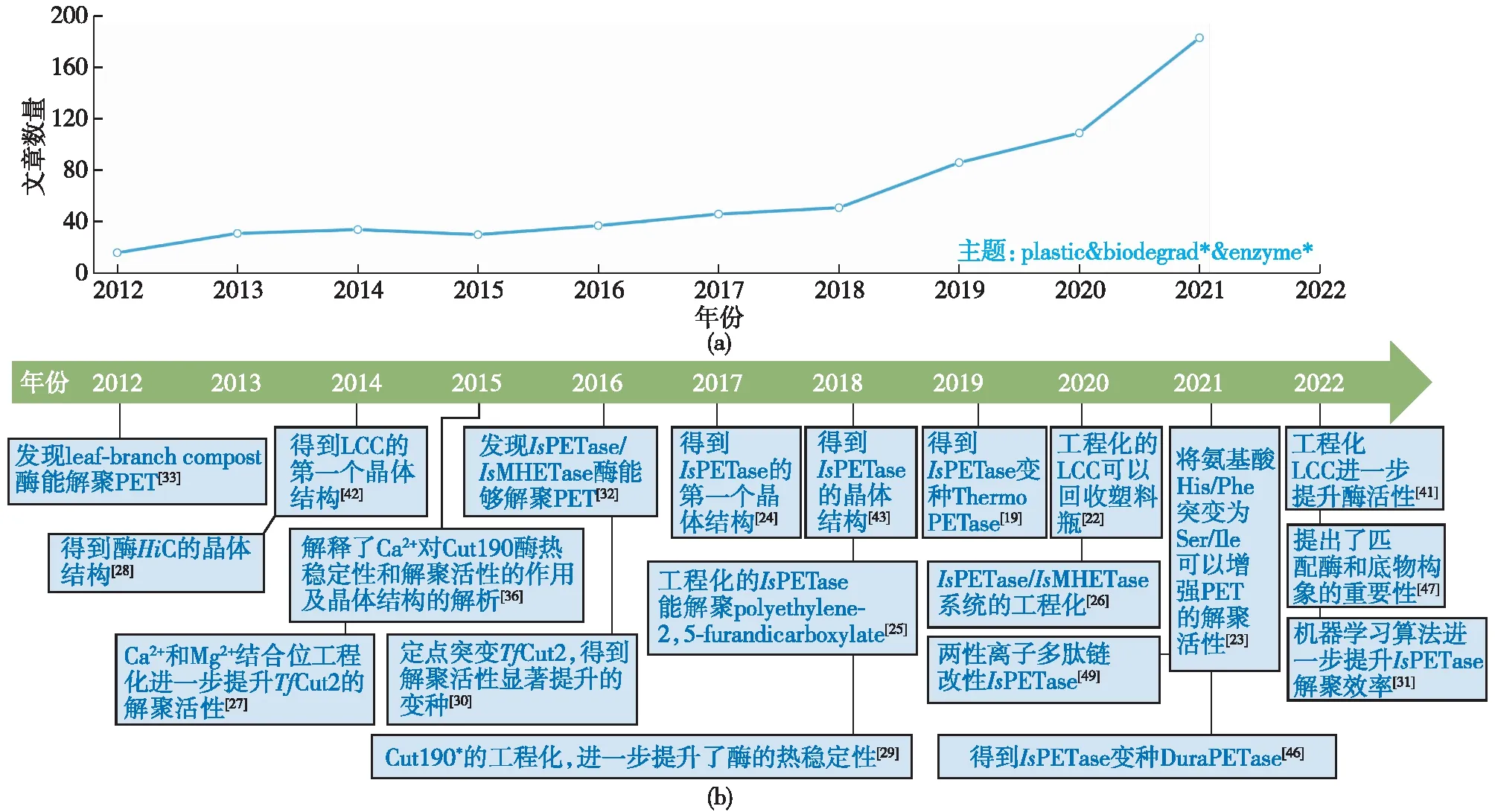
图1 最近十年具有代表性PET解聚酶的鉴别、晶体解析以及工程化的文章发表情况
本文综述了理论计算在PET塑料生物酶解聚研究中的应用进展,探讨了相关研究中的不足与难点,并尝试提出下一步的研究重点,以期理论计算在废弃PET塑料乃至其他废弃聚酯塑料的解聚再利用中发挥更大的作用。
1 理论计算
理论计算主要利用计算机模拟分子的结构与行为,进而模拟分子体系的各种物理、化学性质[56-61]。理论计算技术的发展有2个重要的里程碑:第一个里程碑是1998年诺贝尔奖授予了量子化学家Walter Kohn和John Pople,标志着化学不再是纯粹的实验科学,而由此发展出来的密度泛函理论(DFT)方法已经发展成为广大化学家所使用的工具;第二个里程碑是2013年诺贝尔化学奖授予了理论化学家Martin Karplus、Michael Levitt以及Arieh Warshel,确立了多尺度模拟方法在模拟复杂大分子体系尤其是在模拟生物酶参与的催化反应领域的重要地位[62-66]。按模拟精度,理论计算可以分为从头算(ab initial)、DFT、半经验(semi-empirical)以及分子力学(molecular mechanics,MM)方法等,前三者均属于量子力学方法(quantum mechanics,QM)。一般而言,在计算资源相同的情况下,精度越高的方法能够研究的体系越小。当前量子力学方法仅可以研究包含几百个原子的体系,而分子力学方法可以研究包含上千万个原子的超分子体系,覆盖了当前已知的所有生物酶(PET解聚酶含有约160个氨基酸,约4 000个原子)。
采用理论计算方法研究PET解聚酶催化解聚机制的常见流程主要包括分子对接、动力学模拟、多尺度模拟和数据分析等(图2)。常用的理论计算方法优缺点总结如下:第一,准确的酶催化机制解析依赖精准的酶三维结构。当前蛋白数据库(PDB)中的酶分子活性中心多不含底物分子。虽然有些蛋白结构中包含了底物类似物,如Han等[24]已经解析出含有PET类似物的IsPETase解聚酶晶体结构,为酶理性设计提供了较好的结构模板,然而需要指出的是,底物类似物与真实底物在结构上存在一定的区别,这就导致两者与酶的结合特征也将有所不同。分子对接技术可以快速且相对准确地预测生物酶与真实底物之间的结合模式,是研究解聚酶催化机制的重要技术之一。第二,酶分子柔性大、构象多,准确模拟PET解聚酶的催化解聚机制需要尽可能多地考虑酶的各种构象。分子动力学模拟可以在原子尺度上描述酶催化体系的动态性质,是研究PET解聚酶催化机制的重要手段,其主要缺点是难以准确描述酶催化反应过程。第三,量子化学计算可以描述解聚酶催化反应过程,构建反应势能剖面,明确主要反应通道与速度控制步骤,得到短寿命中间体的结构、频率和能量信息,而其主要缺点是处理的体系大小有限。第四,多尺度模拟技术结合了量子力学和分子力学的优势,采用量子力学方法描述发生化学反应的关键区域,采用分子力学方法描述活性区域周围的环境,能够有效地研究完整的酶分子的催化过程,已经成为研究酶促反应机制的主要方法之一。

图2 理论计算方法研究PET解聚酶催化解聚机制常见流程
2 理论计算技术在塑料生物酶解聚中的应用进展
2.1 分子对接技术应用进展
目前,已经解析出的PET解聚酶三维结构中多没有真实底物[24,41,43,67],因而PET与其解聚酶之间的相互作用与结合模式是当前研究的热点之一。PET解聚酶与PET二聚体的对接模型见图3。Fecker等[68]利用分子对接技术探索了PET二聚体(2-HE(MHET)2)与解聚酶IsPETase、TfCut2和LCC的结合模式,结果发现,PET与IsPETase结合更稳定的根源在于IsPETase结合位点氨基酸残基组成的独特性,如IsPETase中I181的贡献占全部结合能的12%,而LCC(Val)和TfCut2(Ile)中对应位置的氨基酸贡献较小。Liu等[69]采用分子对接技术揭示IsPETase较宽的底物结合口袋是其能够高效降解PET的重要原因之一。Joo等[43]提出PET四聚体(2-HE(MHET)4)可以与IsPETase平坦疏水表面的一个“L”形浅裂缝完美结合,然而该研究中用到的四聚体皆定为trans-构型,这一“假设”的合理性随后被Wei等[70]所挑战:核磁共振(NMR)分析结果表明常温下PET塑料应多以gauche-构型存在(gauche-与trans-构型的比例约为10∶1)。最近,Guo等[47]通过分子对接、动力学模拟等多种技术阐明了PET的构象(trans-和gauche-构型)组成会对解聚酶的解聚活性产生重要影响。除了PET自身结构之外,解聚酶表面结合位点的结构组成也会影响酶的催化效率。分子对接技术可以确定并筛选出与PET结合的关键氨基酸,有针对地开展突变实验可获得解聚活性明显提升的解聚酶突变体。如Son等[71]以PET四聚体(2-HE(MHET)4)为底物,将分子对接与结构生物信息学相结合,对IsPETase的结合位点进行优化,得到了催化活性提升58倍的酶突变体。除IsPETase外,LCC、esterase和PHL7等解聚酶底物结合位点的结构特征也被解析[72-74]。

图3 PET解聚酶与PET二聚体的对接模型
2.2 分子动力学模拟技术应用进展
分子动力学模拟可以描述PET解聚酶在溶液中的动态变化。Chen等[49]发现,将两性离子多肽链融合到IsPETase的C端可以显著提高解聚酶的催化活性,并利用分子动力学模拟对其分子机制进行了解释:计算结果表明两性离子多肽链的引入造成了底物结合口袋处疏水氨基酸的暴露以及Y87残基中苯环的旋转,进而增强了对底物的亲和力(图4)。分子动力学模拟也可用于解释氨基酸突变引起的PET解聚酶催化活性提升的机制。Pirillo等[75]通过分析突变体在有/无底物2种状态下的分子动力学模拟过程,阐明了突变的氨基酸对周围其他氨基酸残基的形态存在影响,进而对酶催化活性的提升作出了合理的解释。Cui等[46]通过晶体结构和MD模拟对DuraPETase突变体进行分析后发现,多种有利突变之间的协同作用对酶稳定性的提升具有重要影响,同时发现突变形成的“芳香通道”有助于酶与PET底物的结合,所以可通过增强活性位点对高结晶度PET底物的适应性来提高解聚酶的解聚活性。分子动力学模拟不仅能解释实验现象,还能用于指导PET解聚酶的改性。如,Li等[55]将分子动力学模拟与机器学习模型相结合预测了氨基酸修饰对蛋白质热稳定性的影响,成功获得具有高Tm的TfCut2突变体,且在70 ℃时PET解聚效率提升了46.42倍。此外,分子动力学模拟技术在MHET水解酶的机制解析中也发挥了重要的作用。如,Peng等[76]利用分子动力学模拟技术深化了对含盖结构域α/β-水解酶IsMHETase构象动力学的认识,为开发工程化更高解聚效率的解聚酶提供了理论基础。

图4 MD模拟轨迹评估EKylated PETases的结构稳定性[49]
2.3 量子化学计算技术应用进展
量子化学计算(包括DFT方法等)能够准确描述体系的化学变化,是当前研究酶催化反应的常见技术之一[77-79]。Hu等[53]利用DFT方法研究了单核金属蛋白酶(NEP)和丝氨酸蛋白酶(CLE)解聚PET的分子机制后发现:这2种酶解聚PET的机制完全不同,其中,单核金属蛋白酶可以利用金属结合的羟基进攻机制(MH)催化PET解聚——酶活性中心锌原子激活水分子,引发亲核进攻反应,继而完成整个催化过程;丝氨酸解聚酶则通过催化三联体(Ser85-His180-Asp165)主导的酰化和脱酰化2个连续的反应过程催化PET解聚。DFT计算结果证实单核金属蛋白酶(38.09 kJ/mol)催化PET的解聚效率比丝氨酸解聚酶(93.77 kJ/mol)高,表明单核金属蛋白酶金属活性中心锌原子的亲核活化作用比丝氨酸解聚酶催化三联体提供的氢键网络稳定作用更有效。此外,DFT方法还可以探究酶与底物之间的相互作用。Feng等[51]和Zheng等[54]应用DFT方法分别研究了解聚酶IsPETase、LCCICCG与底物之间的扰动和相互作用能(图5),阐明了酶催化PET解聚过程中酶自身结构的扰动能变化以及与底物之间相互作用的特征,为合理设计更高催化效率的PET解聚酶提供了思路。该研究提出了决速步比值ΔEint/ΔEdis的概念:对于同样的反应过程,比值越大意味着酶进化得越完美、突变潜力越小。他们发现,LCCICCG的决速步比值虽然较高(0.90),但仍有继续提升的空间。尽管Boneta等[52]基于实验结果认为,LCCICCG突变体水解酶的高活性主要归因于温度因素,但Lu等[31]通过机器学习方法突变得到的FAST-PETase在50 ℃下对PET的解聚效率要高于LCC和LCCICCM在70 ℃下的解聚效率,说明氨基酸突变引起酶内部结构相互作用力(氢键、盐桥等)的改变也可以在一定程度上提高酶的解聚效率。因此,通过改变酶自身结构以及与底物之间的相互作用力也是提升PET解聚酶解聚效率的途径之一。

红色箭头表示活性位点氨基酸的过渡态与反应物/中间体之间的扰动能差;黄色箭头为底物的扰动能差;绿色箭头为过渡态与反应物/中间体的相互作用能差;黑色箭头表示计算出的能垒
2.4 多尺度模拟技术应用进展
多尺度模拟已经成为研究塑料解聚酶促反应的有效手段。Jerves等[48]采用伞形抽样方法在PBE/MM MD水平下研究了解聚酶IsPETase解聚PET的机制后指出,IsPETase解聚过程包括酰化和脱酰化2个阶段,对应的能垒分别为83.72和63.21 kJ/mol。Boneta等[52]采用伞形抽样方法在AM1/MM MD水平下模拟发现:IsPETase和LCC催化PET解聚需要经过4个协同的反应过程,即丝氨酸引起的亲核进攻反应、底物的C—O键断裂、水分子引发的亲核进攻反应、酰化酶中间体的C—O键断裂。根据LCCICCG和IsPETase能垒和水解反应热力学的对比结果可以认为,LCCICCG较高的解聚效率主要归因于高温下酶与底物亲核进攻反应更容易发生,并通过对比解聚酶IsPETase和LCCICCG催化PET二聚体(2-HE(MHET)2)反应过程中的结构变化,揭示了2种重要PET解聚酶之间的差异性。此外,Chen等[80]采用QM/MM水平下的Metadynamics方法也证实了T.fusca角质酶催化解聚机制的酰化和脱酰化2个半反应可以进一步划分为4个协同的反应过程。
酶催化单分子实验(同一个酶分子催化同一种底物)测得的酶催化周转速率常数不是一个恒定的值,而是对应着较长的时间范围(10-3~102s)[81-82],这意味着宏观实验测得的恒定催化速率(kcat值)只是多个酶分子催化反应系综平均后的结果[23,32,44-45]。根据艾林方程,如此大的波动意味着同一酶分子所处的状态不同时,其催化同一种底物对应的能垒差值可达25.12 kJ/mol。考虑多个酶分子构象,采用高水平多尺度模拟方法将能在一定程度上体现这一波动。Feng等[51]和Zheng等[54]从长时间动力学中随机选取20个解聚酶IsPETase和LCC构象,在M06-2X/6-311G(d,p)∥CHARMM高精度计算水平下获得了相关的能量信息(图6),计算结果表明IsPETase和LCC解聚PET二聚体时的决速步的能垒波动范围分别为51.07~149.02和50.23~102.56 kJ/mol。通过采用玻尔兹曼平均方法进一步估算了酶催化反应的能垒(58.19和56.93 kJ/mol),结果与实验值吻合较好。通过构建活性区域关键特征(电荷、结构)与反应能垒之间的线性关系,确定了影响酶解聚效率的关键参数。此外,多尺度计算模拟PET解聚酶的催化反应过程将有助于高效解聚酶的理性设计。Jerves等[48]深入分析了决速步反应物和过渡态的电荷分布后发现,带电残基Asp83、Asp89和Asp159会提升PET解聚反应能垒,因而提出将三者突变为中性氨基酸可能会提升IsPETase的解聚效率。

图6 IsPETase催化PET的解聚过程(a)[51]以及 IsPETase和LCC催化PET解聚反应的能垒(b)[51,54]
3 结论与展望
随着环境中塑料废弃物呈指数倍增加,对塑料废弃物的高效解聚与回收再利用已迫在眉睫。理论计算技术作为一项强有力的研究手段,可以获得酶分子与底物的结合方式、酶分子构象变化、关键氨基酸对酶催化性质的影响等信息,已经在PET塑料生物酶解聚机制挖掘、高效解聚酶设计中发挥了一定的作用。然而,相关研究依然处于相对初级的阶段,仍有许多关键问题待解决:1)PET塑料分子本身结构复杂,存在结晶区、无定形区等,已有研究表明解聚酶对不同结晶度的塑料的解聚效率有明显区别。在当前理论计算研究中,PET塑料多采用小分子寡聚体(如二聚体、四聚体)模型化合物代替,难以准确描述生物酶在塑料超分子表面的吸附和催化行为。2)解聚酶催化全过程模拟研究尚存不足,下一步可以采用统一的计算水平研究解聚酶吸附在PET塑料表面的结合全过程、催化全过程、产物释放全过程。注重探索结合全过程、产物释放全过程是否需要跨越能垒,从酶催化全过程的角度判断影响酶催化速率的关键步骤,从而有针对性地发展酶突变体快速筛选及预测技术。3)在现有服务器计算能力下,理论计算技术在面向超大体系时的计算模拟效率依然有待提升。相信随着大数据技术、人工智能等方法的不断深化与普及,其与理论计算技术的深度结合也必将深化理论计算在塑料生物酶解聚中的应用,为我国塑料废弃物的回收利用做出更大贡献。
