分子动力学模拟在C/SiC材料虚拟试验中的应用与展望
2022-08-05侯传涛朱元夫刘宝瑞吴振强
李 尧,侯传涛, 朱元夫,任 方,刘宝瑞,吴振强
(1.北京强度环境研究所 可靠性与环境技术重点实验室, 北京 100076;2.北京强度环境研究所, 北京 100076)
0 引言
临近空间、可重复使用等飞行器在高速飞行时面临严酷的气动力、热和氧环境,其热防护结构需要同时满足耐高温、气动维形、高承载等要求。近年来, 随着具有耐高温、高比强度和高韧性特点的陶瓷基复合材料制备工艺逐渐成熟,高速飞行器热防护结构研制在“冷热相分离”结构的基础上,一些关键部位逐步采用“防热承载一体化”结构。这些新材料、新型结构的应用使飞行器性能指标得到了提升,但同时给地面验证和评估试验带来困难。一方面,更加严酷高温以及复杂的多物理场耦合环境,使试验环境模拟难度大、成本高;另一方面,温度、热流、应变以及损伤失效参数测试变得更为困难。
近年来,材料结构性能虚拟试验技术逐渐受到关注并得到应用,不仅是对真实试验的补充,而且逐步成为评估和验证材料结构性能的新的技术手段和发展方向。以C/SiC为代表的新一代陶瓷基复合材料优异的综合力学性能来自于多尺度结构设计,其内部包含亚微米厚度的界面层、数十微米直径的碳纤维、毫米尺寸的纤维束以及SiC基体。此外,制备工艺中不可避免产生的大量裂纹、孔隙等缺陷,使其宏观力学行为与传统金属材料显著不同。国内外学者针对C/SiC材料开展了大量多尺度建模与分析工作,能够较准确预示室温下的力学行为。然而,这些多尺度分析多基于有限元方法,其预示精度依赖于强度准则和材料参数的选取。对高温、氧环境下C/SiC材料力学行为机理和规律的认识比较初步,材料参数难以获取、失效准则缺失。因此,C/SiC材料和结构的虚拟试验仍然面临挑战。
分子动力学方法是一种通过模拟构成材料的原子运动,自下而上地预测材料性能及力学行为的方法;但是由于考虑所有组成原子自由度,计算量十分庞大。随着高性能计算、人工智能等技术发展,分子动力学计算能力得到显著提升,应用场景不断丰富,对解决C/SiC虚拟试验的瓶颈问题,展现出较大潜力。本文将首先介绍分子动力学的基本原理,然后对在C/SiC材料模拟方面的进展进行简要综述,最后对未来的应用场景和发展方向进行展望。
1 分子动力学方法介绍
分子动力学有着坚实的理论基础并在金属材料体系中得到大量应用。近年来,结合机器学习等技术,分子动力学模型和算法也在不断发展。本章将从基本原理和发展趋势两个方面对分子动力学方法进行介绍。
1.1 基本原理
微观尺度上,通常认为由原子或分子运动起主导作用,常用于分析固体形成的微结构和缺陷,如:位错、晶界、多相等,发展出晶格动力学、位错动力学等理论分析方法,以及分子动力学、蒙特卡洛等模拟方法。分子动力学不仅模拟体系的能量,还可以获得每一时刻原子的受力,在力学行为研究方面相较蒙特卡洛模拟具有一定优势。分子动力学以统计力学为理论基础,忽略原子核和电子的量子效应,将原子核作为经典粒子,用经典的牛顿运动方程描述组成系统的原子运动过程。式(1)中的,和分别代表时刻第个原子的位置、质量以及受力。体系的总势能()可以通过力场或势函数计算获得,利用式(2)可以计算新的位置处的原子受力。
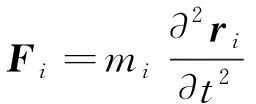
(1)
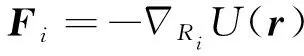
(2)
分子动力学可以计算平衡态系统的结构和能量,以及非平衡态系统动态演化过程中原子的力与运动,例如变形、相变、化学反应等。对原子与原子间相互作用的描述建立在力场(Force field)或势函数之上:在给定的初始条件下,首先利用力场或势函数计算系统的势能();然后求解每个原子受力并计算原子的运动过程,获得新的原子位置和原子速度矢量;重复这一过程,最终得到体系内原子位置随时间的演变过程。相互作用原子的种类、所处的化学环境等不同,力场或势函数对各种原子定义也不同,并采用不同的力常数和经验势参数来描述成键、非键等相互作用。针对不同体系已经形成了Len-nard-Jones势、Tersorff势、SW势、Morse势、嵌入原子势(Embedded Atom Method)、CHARMM势、AMBER势等经验势函数。经验势参数和力常数可以依据材料物性的实验结果获得,或者通过拟合量子力学计算结果获得。
目前,根据研究领域和对象的不同,分子动力学模拟已开发了LAMMPS、GROMACS、NAMD等计算软件,以及OVTIO、VMD等可视化软件,基于高性能计算可实现上亿原子体系的模拟。
1.2 发展趋势
现实中很多现象跨越多个时间和空间尺度,例如,固体变形直至破坏的过程,跨越了从原子结构到宏观结构的多个尺度,是一种复杂的多尺度现象。为了更大的模拟,基于分子动力学的多尺度模拟的研究也在兴起,主要以粗粒化(Coarse Grained)分子动力学方法、桥域法(Bridging domain Method)等多尺度方法为代表。粗粒化分子动力学方法是通过联合原子来构建模型,例如将质量较小的原子附着在重原子上,形成具有新的范德华半径和质量的联合原子,而多个联合原子可以更进一步简化为一个粒子以降低体系的自由度。粗粒化模型简化了部分结构细节,使总粒子数目得到降低,因此所能模拟的空间尺度可增加至原来的几倍,时间尺度提高到原来的几十倍。桥域方法代表多尺度方法中的一类重要方法,将研究区域分为不同区域,采用不同尺度方法进行计算,在区域的边界处进行信息交换。针对固体破坏问题建立的MAAD(Macroscopic, Atomis-tic, Abinitio Dynamics)方法认为对于固体的破坏总是从局部发生,所以可以在局部建立小尺度模型,而在其他地方建立大尺度模型:例如在裂纹极端采用紧束缚模型模拟原子键的破坏,在周围采用利用分子动力学模拟处理裂纹面,而在更远的地方使用连续介质模拟来模拟断裂过程。耦合QM/MM(Quantum Mechnics/Molecular Mechanics)方法是一种结合量子力学的计算精度和分子动力学计算速度的原子模拟方法,将系统分为电子性质重要的区域用量子力学计算而其他区域采用经典描述。类似地还发展出了分子动力学与有限元耦合方法等。经过多年的发展加深了金属材料断裂破坏的认识,并推动了制备工艺的进步和新合金材料的设计,例如高强高韧的纳米孪晶、高温超合金、高熵合金等。
2 C/SiC材料的模拟
陶瓷基复合材料是由纤维、基体、界面层等组分复合形成的多相非均质材料,结构本身具有多尺度特征,还包含大量的大大小小的孔隙和缺陷。目前,陶瓷基复合材料模拟研究仍以有限元方法为主,针对微观、细观、宏观结构分别建模计算,而不同尺度间信息可采用传递、协同、并发等耦合方式。其中,以代表性体积单元方法(Representative Volume Element)和通用单胞模型方法(Generalized Method of Cells)应用最为广泛。代表性体积单元方法的主要思路是选取合适区域建立细观结构模型作为代表性体积单元,通过有限元计算获取应力、应变场量,最后通过均匀化方法对细观场量进行处理,得到宏观材料的应力-应变关系。美国国家航空航天局(NASA)针对复合材料结构多尺度仿真分析提出的通用单胞模型方法,具有宏、细观结构统一的特点,在细观尺度上,进行简化近似并通过解析求解获得细观应力和应变场,而宏观尺度上利用细观均匀化结果进行数值求解,可用于复合材料结构在复杂载荷下的计算分析。然而,有限元计算依赖于微/细观材料组分物理性能的准确获取,同时难以直接模拟氧化、烧蚀等化学反应过程。
近年来,分子动力学模拟研究陶瓷基复合材料物性、化学反应过程的工作不断涌现,对获取陶瓷基复合材料关键力学性能提供了新的途径。下面将通过几个例子介绍分子动力学在C/SiC材料模拟方面的近期研究进展。
2.1 界面相微结构建模
陶瓷基复合材料中亚微米厚度的界面层对其宏观性能影响巨大。热解碳(Pyrolytic Carbon,PyC)是C/C、C/SiC、SiC/SiC等最为常用的界面相材料之一,但由于难以获得较大块体状的热解碳材料,目前对于其力学性能的实验研究较少,且结果分散性较大。为此,许多研究者从原子结构角度出发,对热解碳的微观模型进行了深入研究。2003年,西北工业大学Ye等基于对C/C复合材料中的热解碳结构、模量、强度等性能开展了分子动力学模拟研究。2012年,Leyssale等采用反向蒙特卡洛(RMC)方法通过对HRTEM图像数据处理、三维重构,建立了热解碳的原子构型,如图1 (a)和(b)所示;并且模拟得到纳米尺度热解碳在的弹性常数,虽然与微米级界面相的力学性能相比有一定差别,但是对于认知热解碳微观结构与力学性能之间的关系具有重要意义。2019年,中国科技大学Chen等利用反应力场模拟了热解碳在碳纤维表面的初始沉积生长过程,如图1(c)所示。2020年,Chen等根据沉积的结构设计了代表性的孔洞结构,图1(d)所示,并提出一种便捷的热解碳建模方法:首先在多层石墨烯片层中随机填充该孔洞结构,然后弛豫得到热解碳原子构型;利用获得的模型,研究了热解碳在拉、压和剪切等载荷作用下的微观力学行为。这些工作从原子尺度加深了研究人员对热解碳结构和物性的认识,对于进一步理解陶瓷基复合材料的界面性能以及断裂、脱粘等失效过程具有重要启发。
(a) HRTEM实验数据三维重构图像
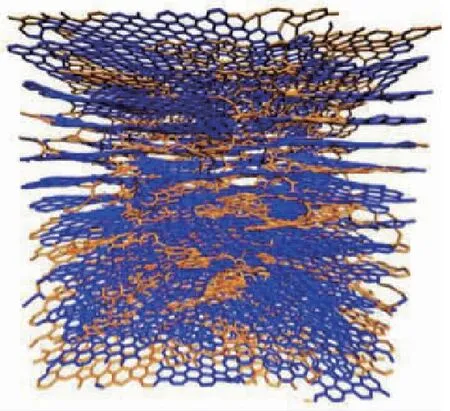
(b) 基于HRTEM图像构建的热解碳原子构型

(c) 热解碳沉积过程模拟
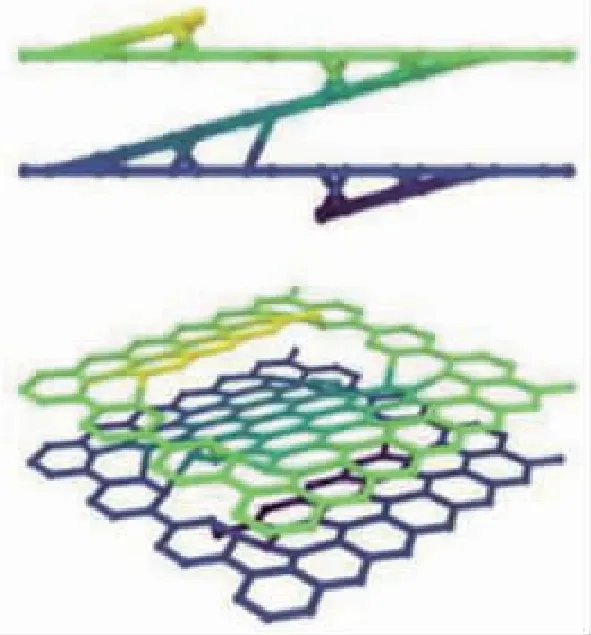
(d) 热解碳中的代表性孔洞结构图1 热解碳原子建模[8-10]Fig.1 Atomic structures of pyrolytic carbon[8-10]
2.2 氧化反应过程模拟
C/SiC复合材料结构在高温环境下长时间使用过程中,氧化是结构损伤失效的重要因素。C/SiC的氧化过程极为复杂,伴随着纤维氧化烧蚀、基体裂纹闭合等多种过程,并且这些过程受温度控制和影响。目前,基于反应和扩展理论,对于C/SiC的氧化反应机理有了定性的认识,但对氧化反应过程定量分析研究仍然处于初步阶段,氧气扩散速率、氧化反应速率等关键参数仍然缺失。
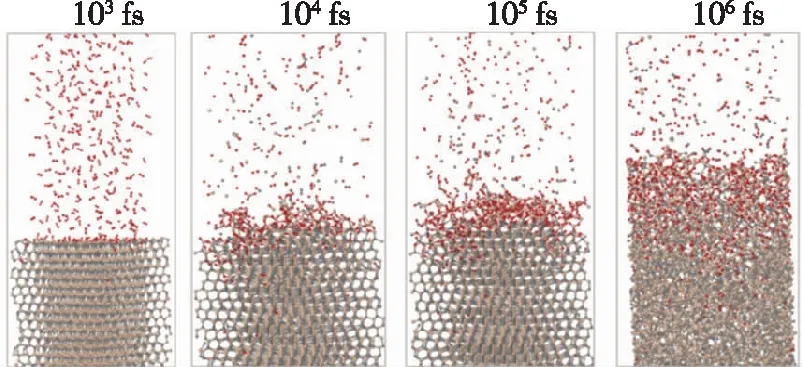
(a) 在1 100 ℃下SiC(0001)表面的氧化过程
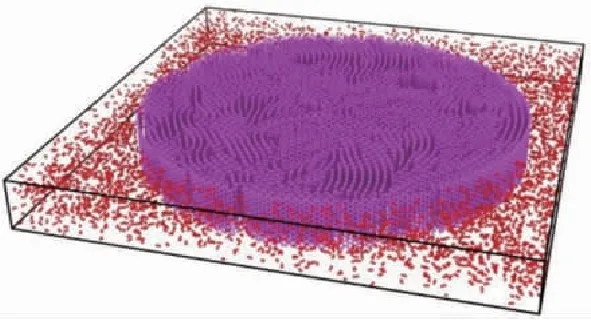
(b) 处于氧气环境中的碳纤维原子结构模型
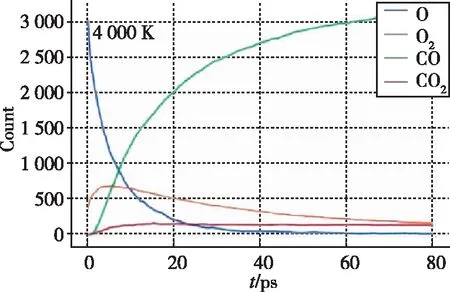
(c) 碳纤维氧化过程中CO分子、O原子和CO2分子数目随时间的变化图2 氧化过程模拟 [14,16]Fig.2 Simulation of oxidation processes [14,16]
2.3 力学性能仿真预示
近年来,学者们针对陶瓷、界面相等组分力学性能以及复合材料微观力学行为开展了相关研究。2019年,Sun等通过分子动力学模拟研究了高温湿氧环境对于Si薄膜中裂纹扩展的影响,如图3(a)所示。2015年,李丽丽等基于Tersoff作用势,通过分子动力学计算研究了无定型碳界面层厚度对SiC纳米纤维/SiC复合材料的断裂力学行为的影响,发现界面层厚度增加会降低纤维的应力集中系数,提高断裂能,使裂纹穿透纤维的脆断模式转变为纤维拔出失效模式,起到增强补韧的效果,如图3(b)所示。图3(c)展示了 Zhang 等理论和实验结合研究了热解碳微柱在拉伸和压缩下的强度性能。2019年,Zhou等通过分子动力学模拟研究了CVI工艺C/C复合材料中热解碳/碳纤维在拉伸、剪切等载荷下的界面力学性质,将获得的界面剪切模量、强度等参数带入有限元模型的内聚力单元中,进行了纤维拔出的有限元仿真分析,如图3(d)所示。
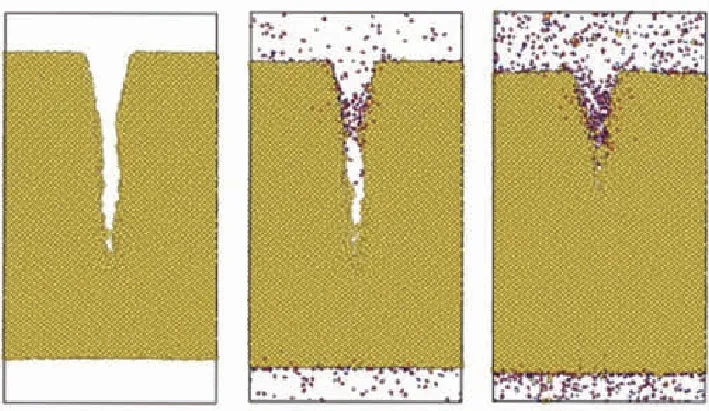
(a) 高温下Si薄膜在真空、氧气和水氧环境中的裂纹扩展
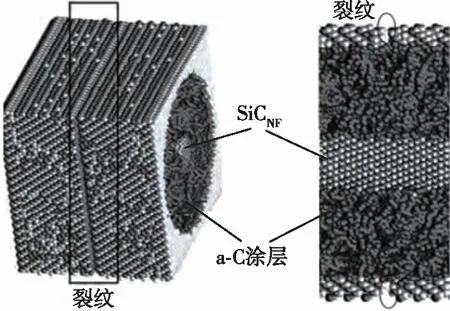
(b) 预置基体裂纹的SiC纤维/无定型C过渡层/SiC复合材料模型
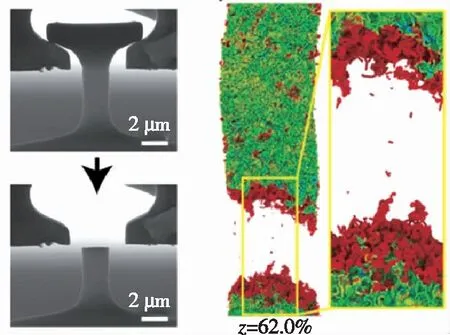
(c) 热解碳微柱单轴拉伸实验和模拟
(d) 热解碳/碳纤维界面的一种典型失效过程图3 力学行为及性能研究[17-20]Fig.3 Molecular dynamics studies on mechanical behaviors and properties [17-20]
3 总论与展望
分子动力学方法在C/SiC材料微结构预测、氧化反应模拟,损伤失效机制、化学反应等方面已经开展了一些探索性研究。本文认为分子动力学在以下几个方面仍具有较大的应用潜力:
(1)支撑材料C/SiC工艺改进以及新材料的研制
分子动力学模拟结合电镜、Raman、CT等测试手段,可以加深对复合材料关键组分,例如界面层的微结构形成及演化规律的认识,帮助建立其微观和宏观性能间关系,实现对材料设计和性能调控。此外,基于分子动力学的反应过程模拟可以帮助优化材料制备工艺参数。对于新材料的研制,可以通过分子动力学进行材料性能预测、先进行初步的筛选,排除错误选项,减少实验工作量,加速研制过程。
(2)支撑极端复杂环境下结构失效分析
对于难以开展试验或测试的极端复杂环境,可以通过分子动力学模拟获得给定载荷环境下材料的应力应变行为和破坏强度,拟合应力应变曲线便可以获得本构模型参数。对于氧化过程,可以类似地拟合出氧化反应速率、氧化损伤因子等参数。这些模型和参数可以直接用于结构的变形及失效分析,帮助厘清失效机理及传播过程。
(3)支撑结构性能多尺度虚拟试验技术发展
以C/SiC为代表的复合材料结构性能不仅取决于其结构参数,还依赖于材料的微细观特征参数以及在结构上的取向和分布。针对这类材料结构,需要考虑其多尺度的特征,在虚拟试验中建立多尺度分析模型。分子动力学除了可以提供材料和模型参数外,还可以对基于有限元方法多尺度模型进行补充和发展,例如分子动力学和有限元耦合模型的构建。
综上所述,随着分子动力学计算能力提升以及新模型新算法的不断提出,必将对材料结构虚拟试验技术的发展带来新的机遇。
