Activation of natural killer T cells contributes to Th1 bias in the murine liver after 14 d of ethinylestradiol exposure
2022-07-30ZouMZKongWCCaiXingMTYuZXChenZhangLYWangXZ
Zou MZ, Kong WC, Cai H, Xing MT, Yu ZX, Chen X, Zhang LY, Wang XZ
Abstract
Key Words: Natural killer T cell; Th1/Th2; IFN-γ; Estrogen; Cholestasis
lNTRODUCTlON
Cholestasis is a mild and common phenomenon during liver diseases but also a crucial triggering element of severe hepatopathy, such as fibrosis, cirrhosis, and hepatic venous thrombosis[1]. Both estrogen and oral contraceptives (OCs) can elicit intrahepatic cholestasis (IHC), whose effects manifest as retention of toxic bile acids in the liver and circulation[2]. 17α-ethinylestradiol (EE) is used as a model drug to induce murine IHC. As the predominant component of OCs and hormone replacement therapy,the clinical counterpart of EE-induced IHC includes women who are taking OCs, postmenopausal replacement therapy and susceptible pregnant women. Sex steroids may lead to alterations in bile components and the elevation of total bile acids (TBAs), which may induce apoptosis and oxidative stress and thus have harmful effects on hepatocytes and other organs[3]. In clinical studies, administration of EE to both males and females leads to increased serum TBA levels and decreased clearance of sulfobromophthalein[4]. In murine studies, the mechanisms involve the activation of AMP-activated protein kinase[5] and estrogen receptor α[6], the inhibition of farnesoid X receptor (FXR), bile acid transporters, bile acid synthase and metabolism enzymes, and inflammatory reactions[7-9]. The first-line therapy ursodeoxycholic acid (UDCA) for cholestatic diseases is a naturally hydrophilic bile acid,although its potency is limited, as approximately 40% of patients are not responsive to UDCA treatment[10].
Among these predisposing factors, the significance of the local immune microenvironment in the liver has been emphasized because sex hormones are immunomodulators that are metabolized in the liver. In a murine model of EE-induced cholestasis, hepatic expression of TNF-α and IL-6 was greatly upregulated[8]. Proinflammatory mediators can affect nuclear receptors and transporters and then cause bile acid equilibrium disorder, increased cytokine secretion, and further exacerbated cholestasis,forming a positive regulation loop[8]. In patients with intrahepatic cholestasis of pregnancy (ICP), an increase in Th1-type cytokines and a decrease in Th2-type cytokines suggest the involvement of proinflammatory and cytotoxic Th1 biases in ICP[11]. However, the impotency of corticosteroids in the treatment of cholestasis indicates that promising therapies or active agents are in heavy demand[12].
Natural killer T (NKT) cells are one of the most abundant lymphocytes in the liver, and their role in cholestasis is noteworthy[13]. NKT cells behave similarly to conventional T cells and function as both effector and regulatory immune cells. Jα18-/-mice are not prone to developing cholestatic liver injury after alpha-naphthylisothiocyanate (ANIT) administration due to the invariant NKT-cell (iNKT cell)knockout-related reduction in cytokines and the restored expression of transporters and bile acid metabolism enzymes[14]. Recent work also demonstrated that IL-4 secreted by type 1 NKT cells (iNKT cells) may inhibit type 2 NKT cells and upregulate immunoregulators, affecting the expression of bile acid nuclear receptors, transporters, and CYP450 enzymes, thus exacerbating triptolide-induced cholestatic liver damage[15]. However, Jα18-/-mice showed aggravation of liver damage after bile duct ligation (BDL) surgery compared with wild-type mice due to the increase in neutrophils, chemokines and cytokines[16]. Moreover, knockout of the bile acid sensor FXR gene increases liver NKT cells and aggravates liver damage, indicating that FXR can regulate the activation of liver NKT cells[17]. Certain antigens that can activate NKT cells exist in the bile of patients with chronic liver diseases[18]. Thus, the aim of the present study was to investigate the effects and mechanisms of NKT cells in a murine model of EE-induced cholestatic hepatotoxicity.
MATERlALS AND METHODS
Reagents
17α-EE (CAS:57-63-6, batch number: E0037, contents > 98.0% (T) high performance liquid chromatography) was purchased from tcichemicals (Shanghai, China). EE was dissolved in an 80% 1,2-propylene glycol solution and diluted with physiological saline to a dosing concentration of 10 mg/kg before the experiment. Anti-CD16/32 antibody (clone: 2.4G2), which was used for blocking before staining, anti-CD3e-FITC antibody (clone: 145-2C11), anti-CD49b-APC antibody (clone: DX5), leukocyte activation cocktail with BD GolgiPlugTM, anti-IFN-γ-PE antibody (clone: XMG1.2), and anti-IL-4-PE antibody(clone: 11B11) were purchased from BD Pharmingen (San Diego, CA, United States).
Animals and treatment
Male C57BL/6J mice (6-8 wk of age and 18-20 g of weight) were obtained from SLAC Laboratory Animal Co., Ltd. (Shanghai, China). iNKT cell-deficient Jα18-/-mice on a C57BL/6 background (6 to 8 wk old) were kindly provided by Dr. Li Bai (University of Science and Technology of China). All mice were administered physiological saline or EE (10 mg/kg) subcutaneously (s.c.) for 14 continuous d.Each group contained 6 mice. All mice were maintained and bred under controlled conditions(pathogen-free, 22 ± 2 °C, 12:12-h light-dark regular photoperiod) with ad libitum mouse chow and water access. The animals were housed in the laboratory for 1 wk prior to experiments to acclimate. All procedures involved in this study were performed under the Ethical Committee of China Pharmaceutical University and the Laboratory Animal Management Committee of Jiangsu Province guidelines(Approval No.: 2021-10-003).
Nonparenchymal cell isolation and labeling
After perfusing saline solution into the heart to eliminate blood, the mouse liver was minced through a 200-gauge nylon mesh and washed with cold PBS. After centrifugation at 50 × g for 2 min in the crude solution, the separated supernatant was centrifuged for another 10 min at 800 × g. The cell pellets were resuspended in 40% Percoll for centrifugation at 1250 × g for 15 min. After nonparenchymal cell (NPC)isolation from the cell pellets by red blood cell lysis solution (0.15 M NH4Cl and 0.1 mmol/L Na2EDTA)treatment and 2 washes, NPCs were stimulated with leukocyte activation cocktail for 4-5 h (BD Pharmingen).
After stimulation, NPCs were blocked with anti-CD16/32 and then surface-labeled with FITCconjugated anti-mouse CD3e and APC-CD49b antibodies. NPCs were permeabilized with Cytofix/Cytoperm (Becton Dickinson) following the manufacturer’s instructions, and intracellular staining was performed by incubation with IFN-γ or IL-4 antibodies. After each step, unbound antibodies were excluded from the system by centrifugation. Then, the cells were analyzed using a FACSCalibur flow cytometer (Becton Dickinson, Palo Alto, CA, United States), with 50000 events recorded per tube. The results were processed using FlowJo version 10 software (FlowJo, Ashland, OR, United States). The gating strategy was as follows: first, NPCs were gated by FSC and SSC, followed by the gating of CD3e+or CD3e-cells. CD49b+IFN-γ+, CD49b+IL-4+or CD49b-IFN-γ+, and CD49b-IL-4+cells were then gated and analyzed.
Blood biochemical analysis
Anticoagulant-free serum was collected and then analyzed for the levels of alkaline phosphatase (ALP),total bile acid (TBA), alanine transaminase (ALT), and aspartate transaminase (AST) using the ALP,ALT, and AST quantification kit (Whitman Biotech, Nanjing, China) and TBA quantification kit(Jiancheng Bioengineering Institute, Nanjing, China).
Histopathology and immunohistochemistry
The liver lobules were fixed, paraffin embedded, sectioned, and stained with hematoxylin and eosin for histopathological examination. Slides were coded, randomized, and assessed by pathologists who,during the evaluation of the slides, were blinded to the treatment groups. Scoring for liver injury was conducted according to the following criteria: no hepatocellular necrosis, proliferation of pseudocholangiolar duct, or inflammatory cell infiltration, 0; mild, 1; moderate, 2; severe, 3. The other sections weresubjected to IHC for semiquantification of the expression of toll-like receptor (TLR) 9 (Novus, 26C593.2,dilution percentage: 1:200).

Table 1 The primer sequence used for real-time polymerase chain reaction in mice
RNA extraction and real-time polymerase chain reaction
RNA was extracted from liver sections by TRIzol reagent (Vazyme Biotech, Nanjing, China). Isolated RNA was processed by the HiScriptTMQ RT SuperMix for quantitative polymerase chain reaction(qPCR) (+ gDNA wiper) kit (Vazyme Biotech) for cDNA synthesis. A 20 μL real-time PCR system was prepared according to the manufacturer’s instructions. The mRNA levels were normalized to the housekeeping geneGapdh. The primer sequences used are shown in Table 1.
Statistical analysis
Values are expressed as the means ± SE. The groups were evaluated using Student’sttest. APvalue <0.05 was considered statistically significant.
RESULTS
EE promotes hepatic NKT-cell proliferation along with biased Th1 cytokine production
Increased activation of NKT cells, T cells, or NK cells has been reported to contribute to the pathophysiology of cholestasis[15,19,20]. Here, we measured and analyzed their percentages and Th1/Th2 cytokine production to evaluate their activation. Hepatic NKT cells, CD3+T cells (excluding NKT cells) and NK cells were defined as CD3e+CD49b+(Figure 1A upper right), CD3e+CD49b-(Figure 1A lower right), and CD3e-CD49b+(Figure 1A upper left), respectively. After EE administration,the percentage of hepatic NKT cells expanded more than 3-fold compared with the control group, while the frequencies of CD3+T cells and NK cells showed no significant changes (Figure 1A-D). Both IFN-γ and IL-4 secreted from NKT cells increased, which further indicated NKT-cell activation. Moreover, the upregulated Th1/Th2 ratios indicated that a skewed hepatic Th1 immune response was due to NKT cells (Figure 1E-I). Compared with the control group, IL-4 secreted by CD3+T cells and NK cells increased, and their IFN-γ secretion and Th1/Th2 ratios remained unaltered (Figure 1J-Q).
iNKT cells exacerbate EE-induced cholestatic liver damage
For further investigation of the NKT-cell effect, cholestatic liver injury was compared between iNKT cell knockout mice (Jα18-/-mice) and C57BL/6J mice after EE administration. Compared with the control group, EE induced increased levels of ALP, TBA, ALT, and AST in C57BL/6J mice. Compared with C57BL/6J mice, Jα18-/-mice demonstrated significantly lower levels of ALP, TBA, ALT, and AST(Figure 2A-D). Based on the scoring criteria[21], the histopathological and hepatic injury score results showed proliferation of pseudocholangiolar duct (yellow arrows), inflammatory cell infiltration (red arrows) and hepatocyte necrosis (black arrows) after EE treatment, whereas Jα18-/-mice exhibited reduced proliferation of the pseudocholangiolar duct, inflammatory cell infiltration, and necrosis(Figure 2E-G). These results showed that iNKT cell-deficient mice can inhibit the development of cholestatic hepatotoxicity induced by EE, indicating a pathogenic effect exerted by iNKT cells on EEinduced cholestatic liver damage.

Figure 1 Ethinylestradiol promotes hepatic natural killer T cell proliferation along with biased Th1 cytokine production. A: The percentage of hepatic natural killer T (NKT) cells, T cells and NK cells were detected and defined as CD3e+CD49b+ (upper right), CD3e+CD49b- (lower right), CD3e-CD49b+ (upper left), respectively; B: Proportion analysis and comparison of NKT cells; C: Proportion analysis and comparison of T cells; D: Proportion analysis and comparison of NK cells; E and F: Th1 cytokine (IFN-γ) (E) and Th2 cytokine (IL-4) (F) produced by NKT cells and T cells were detected, respectively; G: IFN-γ produced by NKT cells; H: IL-4 produced by NKT cells; I: The ratio of Th1/Th2 by NKT cells; J: IFN-γ produced by T cells; K: IL-4 produced by T cells; L: The ratio of Th1/Th2 by T cells;M and N: Th1 cytokine (IFN-γ) (M) and Th2 cytokine (IL-4) (N) produced by NK cells were detected; O: IFN-γ produced by NK cells; P: IL-4 produced by NK cells; Q:The ratio of Th1/Th2 by NK cells. All values are the means ± SE (n = 4-6). aP < 0.05, cP < 0.001 vs control.

Figure 2 lnvariant natural killer T cells exacerbate ethinylestradiol-induced cholestatic liver damage. A-D: The serum levels of ALP (A), TBA (B),ALT (C) and AST (D); E: Liver sections stained with hematoxylin and eosin (10×); F: Proliferation of pseudocholangiolar duct (yellow arrows); G and H: Inflammatory cell infiltration (G, red arrows) and hepatocyte necrosis (H, black arrows) were compared as pathology scores. All values are the means ± SE (n = 4-6). bP < 0.01, cP< 0.001 vs control; eP < 0.01, fP < 0.001 vs C57BL/6J group.
iNKT cell deficiency lowers the hepatic expression of chemokine/chemokine receptors
CXCR6, the receptor for chemokine CXCL16, is expressed on the surface of several hepatic lymphocytes,including NKT cells. The CXCR6-dependent infiltration of NKT cells into the liver induces enhanced inflammation in a murine model of chronic hepatic damage[22]. The qPCR results showed that EE promoted the hepatic mRNA expression of Cxcr6/Cxcl16 in C57BL/6J mice. Compared with C57BL/6J mice, iNKT cell deficiency significantly suppressed the mRNA expression of Cxcr6/Cxcl16, which may be related to the absence of the need for hepatic recruitment of NKT cells (Figure 3).
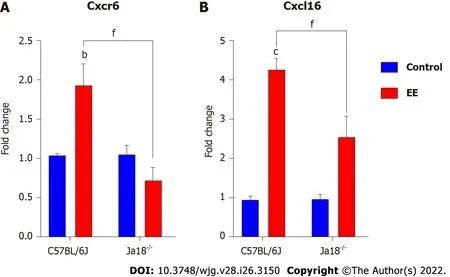
Figure 3 lnvariant natural killer T cell deficiency lowers the hepatic expression of chemokine/chemokine receptors. A and B: Hepatic mRNA expressions of Cxcr6 (A) and Cxcl16 (B) were determined and compared. All values are the means ± SE (n = 4-6). bP < 0.01, cP < 0.001 vs control; fP < 0.001 vs C57BL/6J group.
Absence of iNKT cells alters the expression of TLRs and associated signaling pathways
Accumulating evidence suggests that TLRs contribute to the development of hepatobiliary damage[23].TLRs have also been reported to contribute to the activation and subsequent cytokine production of NKT cells[24]. Intriguingly, iNKT cells have been reported to express certain TLRs, some of which are functional[25]. In the present study, TLRs associated with NKT-cell activation or liver disease were measured and analyzed. The results showed that the mRNA expression levels of Tlr2, Tlr4, Tlr6, Tlr7,and Tlr9 were significantly upregulated after EE administration. Deficiency of iNKT cells significantly downregulated the expression of the above TLRs compared with that in C57BL/6J mice (Figure 4),which suggested that the absence of iNKT cells may influence the expression of TLRs. The hepatic immunohistochemistry results of TLR9 showed a trend similar to that of mRNA (Figure 4F).
TLRs or T cell receptors (TCRs) can stimulate T cells, leading to MAPK signaling pathway activation,which is required for the effects of T cells[26]. iNKT cell activation by TCR stimulation has also been reported to be mediated by ERK and p38 MAPK[27]. In the present study, the expression of both MAPK and PI3K signaling was investigated after EE administration in C57BL/6J and Jα18-/-mice. The hepatic mRNA expression levels of MAPK upstream Ras and Raf as well as Pi3k and its downstream Bad,which might be associated with TLR activation, were increased in C57BL/6J mice after EE administration. The above mRNA levels were remarkably inhibited in Jα18-/-mice compared with C57BL/6J mice, which suggested that the absence of iNKT cells changes the expression of MAPK and PI3K signaling pathways (Figure 5).
Knockout of iNKT cells may inhibit the synthesis of bile acid and weaken the inhibition of hepatocyte polarity
Dysregulated bile acid homeostasis may be attributed to the disorder of bile acid synthesis/metabolism and/or the dysfunction of tight junctions (TJs)[28]. To investigate the possible mechanism of the alleviation of cholestatic liver injury in NKT-cell knockout mice, the levels of CYP450 synthase and TJs were detected. The mRNA levels of Cyp7a1 and Cyp8b1 were upregulated after EE administration in C57BL/6J mice, whereas a nonsignificant difference was shown in Jα18-/-mice. In comparison to C57BL/6J mice, the mRNA expression of Cyp7a1 and Cyp8b1 was inhibited in Jα18-/-mice after EE administration, indicating the relatively reduced synthesis of bile acid (Figure 6A and B). The IHC results showed that, in comparison to the control group, fewer cells positively stained for ZO-1 or Occludin after EE treatment in C57BL/6J mice, and positive cells remained unaltered between the control and EE groups in Jα18-/-mice (Figure 6C and D). These data suggested that knockout of iNKT cells can inhibit bile acid synthase and weaken the inhibition of hepatocyte polarity, thereby reducing cholestatic liver injury.
DlSCUSSlON
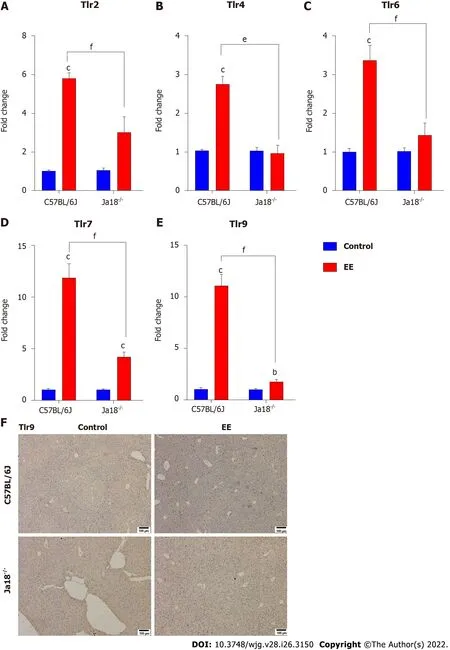
Figure 4 Absence of invariant natural killer T cells alters the expression of toll-like receptors. A-E: Hepatic mRNA levels of Tlr2 (A), Tlr4 (B), Tlr6(C), Tlr7 (D) and Tlr9 (E) were determined and compared; F: Changes in the protein level of TLR9 were determined by immunohistochemistry. All values are the means ± SE (n = 4-6). bP < 0.01, cP < 0.001 vs control; eP < 0.01, fP < 0.001 vs C57BL/6J group.

Figure 5 Absence of invariant natural killer T cells alters the expression of Ras/Raf and Pl3K/Bad signaling pathways. A-D: Hepatic gene expressions of Ras (A), Raf (B), Pi3k (C) and Bad (D) were determined and compared. All values are the means ± SE (n = 4-6), bP < 0.01, cP < 0.001 vs control; eP <0.01, fP < 0.001 vs C57BL/6J group.
As the most common hepatic disorder, the principal pathogenic factors of EE-induced IHC include estrogen and toxic bile acid, both of which are correlated with immune imbalance. Increased activation status of NKT cells, T cells, or NK cells has been reported to contribute to the pathophysiology of cholestatic diseases[15,19,20]. Therefore, the percentages of hepatic NKT cells, CD3+T cells, and NK cells along with their Th1/Th2 cytokine secretion and bias were compared in the present study. Surprisingly,in EE-induced cholestasis, only the NKT-cell percentage and its Th1/Th2 cytokine secretion and ratio significantly increased, indicating their activation and Th1-biased immune response. The percentage of CD3+T cells and NK cells, along with their secretion of Th1 cytokines, remained unaltered. Th2 cytokines from CD3+T cells and NK cells showed a rising trend, which did not seem to affect the Th1/Th2 balance (Figure 1). Our results revealed that both Th1 and Th2 cytokines existed in the liver,consistent with previous studies[14,29], and the Th1 bias was mainly contributed by NKT cells. To further confirm the effect of NKT cells in EE-induced cholestasis, cholestatic liver damage was compared between iNKT cell-deficient mice and C57BL/6J mice after EE administration. Jα18-/-mice showed significantly reduced blood biochemistry parameters, including levels of ALP, TBA, ALT, and AST, and alleviated liver histopathological changes, suggesting that NKT cells have a harmful effect on EE-induced cholestasis. NKT cells have been accepted as a key connection between innate and adaptive immunity. Upon activation, NKT cells can activate and influence most innate and adaptive immune cells by rapid and abundant cytokine production[30,31]. NKT cells, whose role is noteworthy in cholestasis, are mostly abundant in the liver rather than other organs. In the PBC mouse model, NKT cells are highly activated and secrete enormous amounts of IFN-γ. In comparison to mice with PBC,cholangitis and liver inflammation are significantly alleviated in mice with NKT-cell deficiency[32]. Our previous investigations also found that iNKT cells and their cytokines aggravate ANIT- and triptolideinduced cholestasis by disrupting bile acid homeostasis[14,15]. Although NKT cells can be protective by suppressing neutrophils in an extrahepatic cholestasis model[16], our research data suggest the harmful effects of NKT cells in EE-induced cholestasis. The above studies indicate that the role of NKT cells may vary in different diseases and models.
Under physiological conditions, CXCR6/CXCL16 regulate the migration of NKT cells in hepatic sinusoids[33]. Under the early pathologic states of chronic liver damage, they specifically mediate the hepatic accumulation of NKT cells, exacerbating hepatic inflammation and promoting fibrogenesis[22].CXCL16 can be expressed in hepatobiliary tissues of patients with hepatopathy[34] but also in mouse liver sinusoidal endothelial cells triggered by intestinal microorganism-induced bile acids, indicating that its expression is related to hepatobiliary damage[33,35]. Our results showed that the hepatic gene levels of Cxcr6 and Cxcl16 were upregulated (Figure 3). Inflammatory cytokines, such as IFN-γ, which was increased in the present model, are capable of further inducing CXCL16 expression, forming a positive regulation loop[36]. In the present study, iNKT cell deficiency inhibited the mRNA levels of Cxcr6 and Cxcl16, which may be linked to the absence of hepatic infiltration of NKT cells (Figure 3).
Activation of iNKT cells can be caused by direct interaction with TCR or indirect effects of TLR agonists and/or IL-12[37]. TLRs, which are widely expressed in the liver, can stimulate hepatic inflammation and actively participate in the initiation and development of liver damage under pathological conditions[38,39]. Lipopolysaccharide (LPS), a specific TLR4 Ligand, activates hepatic iNKT cells and leads to their secretion of IL-4 within 2 hin vivoor in vitro, demonstrating that TLR4 is expressed and functional in iNKT cells[40]. In alcoholic liver disease, TLR4 inhibition results in drastically reduced levels of hepatic proinflammatory mediators[41]. TLR4 deletion nearly eliminates inflammatory cell infiltration in the liver and hepatocyte injury in a murine model of ischemia reperfusion[42]. In EEinduced cholestatic liver injury, hepatic mRNA levels of Tlr2, 4, 6, 7, and 9 were markedly increased(Figure 4). CD1d knockout mice (lacking NKT cells) exhibit enhanced (> 4-fold) proinflammatory cytokine secretion and higher mRNA levels of TLR4 in the kidney of a nonalcoholic fatty liver disease model[43]. In the present study, iNKT cell deficiency significantly downregulated the expression of the above TLRs compared with C57BL/6J mice (Figure 4), suggesting the effect of iNKT cells on the expression of TLRs. NKT cells have been found to express certain functional TLRs[24]. The IHC results of TLR9 showed a similar trend as the mRNA levels (Figure 4F). In a murine autoimmune hepatitis model induced by concanavalin A, NKT cells infiltrate and are activated, promoted by TLR9 stimulation, thereby leading to aggravated hepatotoxicity[44]. The PI3K/Akt and Ras/Raf/MEK/ERK signaling pathways are considered to be activated and function following inflammation[45], which further upregulates proinflammatory mediators in cholestasis[46]. Our data demonstrated that the mRNA levels of the downstream pathways Ras/Raf and Pi3k/Bad were also upregulated, while iNKT cell deficiency suppressed their upregulation (Figure 5). These results indicated that iNKT cell deletion affected TLRs and their downstream Ras/Raf and PI3K/Bad signaling.
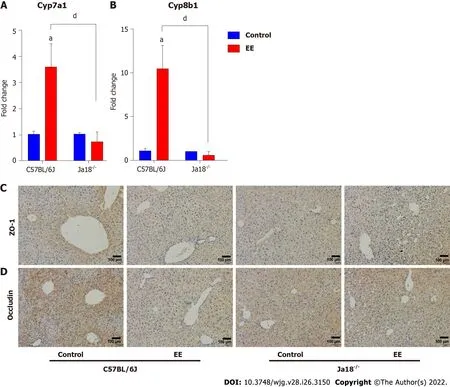
Figure 6 Knockout of invariant natural killer T cells may inhibit the synthesis of bile acid and weaken the inhibition of hepatocyte polarity.A and B: Hepatic gene levels of bile acid synthase Cyp7a1 (A) and Cyp8b1 (B) were determined and compared; C and D: Changes in the protein expressions of tight junction Zo-1 (C) and Occludin (D) were determined by immunohistochemistry. All values are the means ± SE (n = 6), aP < 0.05 vs control; dP < 0.05 vs C57BL/6J group.
Growing numbers of studies have reported that hepatocyte TJs, constituting hepatocyte polarization,play a pivotal role in the maintenance of the epithelial barrier and permeability; therefore, their structural disruption can lead to the leakage of bile components into blood circulation, bile acid homeostasis disorder, and cholestatic liver injury[47]. TJ damage can be found in obstructive jaundice patients[48] and murine cholestatic models of ANIT[28], carbon tetrachloride[49], BDL[50], and EE[51].Inflammation has been associated with structural and functional alterations of hepatic TJs[52]. In the present study, the results showed that iNKT cell-deficient mice ameliorated disorders of bile acid synthesis and TJs (Figure 6) and restored previously deteriorated bile acid homeostasis and hepatocyte barrier function, contributing to the mitigation of EE-induced cholestatic hepatotoxicity.
CONCLUSlON
In EE-induced cholestatic hepatotoxicity, EE promoted hepatic NKT-cell proliferation and activation,which contributed to Th1 cytokine bias and influenced the liver immune microenvironment. EE also upregulated the expression of Cxcr6/Cxcl16, TLRs, and downstream Ras/Rad and PI3K/Bad signaling.Moreover, EE influenced the levels of the bile acid synthase Cyp7a1 and Cyp8b1 and the TJs ZO-1 and Occludin. iNKT cell deficiency significantly alleviated cholestatic liver damage and downregulated the abovementioned signaling pathways, indicating the pathogenic effects of hepatic iNKT cells in EEinduced cholestatic liver damage. It is noteworthy that mouse and human NKT cells share similar functions, including the killing of tumor/infected cells by cytotoxicity and their crucial role in autoimmunity[53]. Therefore, regulation of NKT-cell activation may serve as a potential therapeutic strategy with clinical implications for cholestatic diseases.
ARTlCLE HlGHLlGHTS

ACKNOWLEDGEMENTS
The authors would like to acknowledge Li Bai (University of Science and Technology of China) for kindly providing iNKT cell-deficient (Jα18-/-mice).
FOOTNOTES
Author contributions:Zou MZ and Kong WC contributed equally to the work; Zou MZ, Kong WC, Yu ZX, and Chen X performed the experiments, collected data, and analyzed the data; Xing MT, Zhang LY, and Wang XZ contributed to the guidance of experiments and the final manuscript; Wang XZ and Xing MT designed the study; Wang XZ, Xing MT, Zou MZ and Cai H wrote the manuscript; All the authors have reviewed and approved the final version of the manuscript.
Supported bythe National Natural Science Foundation of China, No. 82073948 and 81703626; and National Innovation and Entrepreneurship Training Program for Undergraduate, No. 202210316040Z.
lnstitutional animal care and use committee statement:All animal experiments involved in this study were performed under the Ethical Committee of China Pharmaceutical University and the Laboratory Animal Management Committee of Jiangsu Province guidelines (Approval No.: 2021-10-003).
Conflict-of-interest statement:All authors have nothing to disclose.
Data sharing statement:No additional data are available.
ARRlVE guidelines statement:The authors have read the ARRIVE guidelines, and the manuscript was prepared and revised according to the ARRIVE guidelines.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:China
ORClD number:Meng-Zhi Zou 0000-0001-5303-7550; Wei-Chao Kong 0000-0001-9083-2609; Heng Cai 0000-0001-6544-2414; Meng-Tao Xing 0000-0003-3816-4214; Zi-Xun Yu 0000-0002-7272-7394; Xin Chen 0000-0002-5330-6543; Lu-Yong Zhang 0000-0003-1164-7371; Xin-Zhi Wang 0000-0001-9624-0102.
S-Editor:Zhang H
L-Editor:A
P-Editor:Guo X
杂志排行

World Journal of Gastroenterology的其它文章
- Involvement of Met receptor pathway in aggressive behavior of colorectal cancer cells induced by parathyroid hormone-related peptide
- Role of gadoxetic acid-enhanced liver magnetic resonance imaging in the evaluation of hepatocellular carcinoma after locoregional treatment
- Clinical implications and mechanism of histopathological growth pattern in colorectal cancer liver metastases
- Bifidobacterium infantis regulates the programmed cell death 1 pathway and immune response in mice with inflammatory bowel disease
- Tumor-feeding artery diameter reduction is associated with improved short-term effect of hepatic arterial infusion chemotherapy plus lenvatinib treatment
- Impact of sodium glucose cotransporter-2 inhibitors on liver steatosis/fibrosis/inflammation and redox balance in non-alcoholic fatty liver disease