BAPTA 受体荧光材料的差异性结构构筑与合成*
2022-07-01成昭,郑蕾,徐玥,何昊
成 昭,郑 蕾,徐 玥,何 昊
(西安医学院 药学院,陕西 西安 710021)
荧光材料与荧光分析方法,因其良好的光学性能、定性及定量分析特性,得到广泛应用。荧光材料光学特性与其分子结构关系密切,进行材料荧光性能调控时,需综合考虑分子刚性、平面性、共轭结构与取代基等影响因素[1,2]。其中,基于平面大π 键结构的刚性分子骨架,进行取代基修饰,对材料的荧光性能影响显著,相关研究工作屡见报道[3-5]。实际工作中,主要依靠给电子、吸电子取代基引入,实现材料的荧光性能调控。其中,给电子基团-OH、-OR、-NR2等,因p-π 共轭作用形成,使电子共轭程度增强、电子云密度增大,材料荧光性能随之增强,一般还伴随着荧光材料的荧光发射波长红移、荧光量子产率增大。当吸电子基团-COOH、-NO2、-X 等被引入分子时,则使电子共轭程度降低、电子云密度减小,材料荧光减弱。
本文设计合成BAPTA 受体荧光材料S15、S16、S17 时,分别在其荧光发色团的杂环结构中引入吸电子取代基4-NO2、合成得到S17,引入给电子取代基N-CH3、得到S16,期望调控S16 与S17 的电子云密度及荧光性能。作为最小的烷基与给电子取代基,甲基的引入不会造成较大空间位阻[6,7],而甲基引入杂环后显著的共轭与超共轭作用,预期将使S17 的荧光性能显著增强,硝基对S16 的影响则反之。此外,综合比较单发色团荧光材料钙绿-1 与相应双发色团材料钙绿-2 的结构及性能[8],发现双发色团致使荧光增强,因此,本文亦合成具有苯并咪唑双发色团的荧光材料S15,以单发色团材料S16 与S17 为对照、研究S15 的荧光性能增效。
1 实验部分
1.1 主要试剂与仪器
邻硝基酚、1-溴-2-氯乙烷、溴乙酸甲酯、邻苯二胺、N-甲基邻苯二胺、4-硝基邻苯二胺、无水K2CO3、还原铁粉,均为化学纯,国药集团化学试剂有限公司;N,N-二甲基甲酰胺(DMF)、甲醇、乙醇、乙酸乙酯、石油醚,均为化学纯,天津市天力化学试剂有限公司。
XT-4 型显微熔点仪(北京泰克);TENSOR T-27型傅里叶变换红外光谱仪(美国布鲁克);AVANCE III 400MHz 型超导核磁共振波谱仪(美国布鲁克);microTOF-QⅡESI-Q-TOF LC/MS/MS 型飞行时间-质联仪(美国布鲁克);LS-55 型荧光分光光度计(美国铂金埃尔默)。
1.2 实验方法
基于前期研究工作中合成得到的荧光材料S11结构,针对材料发色团的荧光基团数目、电子云密度、分子刚性等结构因素进行差异性设计,以邻硝基酚、1-溴-2-氯乙烷、溴乙酸甲酯、邻苯二胺、N-甲基邻苯二胺、4-硝基邻苯二胺等为原料,经6 步合成得到3 种新型BAPTA 受荧光材料1,2-二(2-氨基-5-苯并咪唑基苯氧基)乙烷-N,N,N',N'-四乙酸甲酯(S15)、2-[2-(2-氨基苯氧基)乙氧基]-4-(N-甲基苯并咪唑基)苯胺-N,N,N',N'-四乙酸甲酯(S16)、2-[2-(2-氨基苯氧基)乙氧基]-4-(4-硝基苯并咪唑基)苯胺-N,N,N',N'-四乙酸甲酯(S17),合成路线见图1、2。
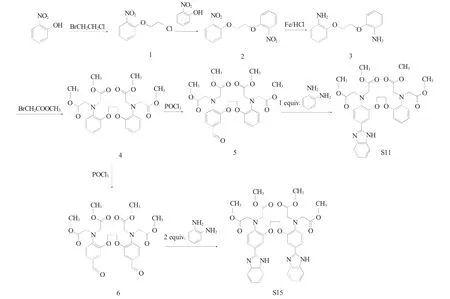
图1 BAPTA 受体荧光材料S15 的合成路线Fig.1 Synthetic route of BAPTA fluorescent material S15

图2 BAPTA 受体荧光材料S16、S17 的合成路线Fig.2 Synthetic route of BAPTA fluorescent materials S16 and S17
1.3 1,2-二(2-氨基-5-苯并咪唑基苯氧基)乙烷-N,N,N',N'-四乙酸甲酯(S15)的合成
1.3.1 2-(2-氯乙氧基)-硝基苯(化合物1)的合成 将邻硝基酚(1.39g, 0.01mol)、1-溴-2-氯乙烷(4.30g,0.03mol)与无水K2CO3(2.07g, 0.015mol)均匀分散于DMF(8mL)中,120℃反应5h。待反应结束、冷却,加入20mL 乙酸乙酯稀释、水洗(10mL×3)、旋干,以甲醇-水重结晶,得到黄色固体1,产率93%,m.p.36~37℃。1H NMR(CDCl3,400MHz),δ:7.85(d, J=8Hz,1H, ArH),7.53~7.57(m, 1H, ArH),7.04~7.23(m,2H, ArH),4.40(t, J=12Hz, 2H, -CH2O-),3.86(t, J=12Hz, 2H, -CH2Cl)。IR(KBr),ν,cm-1:2972.34,2926.51,2875.20(νC-H);1608.27,1484.98(νC=C);1522.27,1343.29(ν-NO2);1247.30,1026.00(νC-O-C);745.61,667.76(δ=C-H)。
1.3.2 1,2-二(2-硝基苯氧基)乙烷(化合物2)的合成 将化合物1(2.01g, 0.01mol)、邻硝基酚(1.39g,0.01mol)与无水K2CO3(2.50g, 0.018mol)均匀分散于DMF(10mL)中,140℃反应4h。待反应结束、冷却,加入20mL 冰水,经抽滤、水洗(10mL×3)、甲醇-水重结晶,得到黄色固体2,产率为95%,m.p.168~169℃。1H NMR(CDCl3,400MHz),δ:7.83(d, J=8Hz,2H, ArH),7.57(t, J=8Hz, 2H, ArH),7.24(t, J=8Hz,2H, ArH),7.08(t, J=8Hz, 2H, ArH),4.54(s, 4H,-OCH2CH2O-)。IR(KBr),ν,cm-1:3051.66(ν=C-H);2956.13,2931.93,2873.61,2760.34(νC-H);1606.92,1582.83,1484.65(νC=C);1518.56,1359.96(νNO2);1278.08,1090.66(νC-O);744.19,671.99(δ=C-H)。
1.3.3 1,2-二(2-氨基苯氧基)乙烷(化合物3)的合成 将浓HCl(0.2mL)、无水乙醇(10mL)、还原铁粉(3.36g, 0.06mol)混合均匀,80℃反应30min,再向其中分批加入化合物2(3.04g, 0.01mol),80℃反应4h。待反应结束,向其中趁热加入15% KOH-乙醇、至pH 值为7~8,滤除铁粉,向滤液中加入6mol·L-1H2SO4,将硫酸盐沉淀过滤并溶于热水,再加入饱和NaOH 至pH 值为9~10,冷却、抽滤、甲醇重结晶,得到白色固体3,产率88%,m.p.116~117℃。1H NMR(CDCl3,400MHz),δ:6.91~7.04(m, 8H, Ar-H),4.36(s, 4H, -OCH2CH2O-),3.82(s, 4H, 2×-NH2)。IR(KBr),ν,cm-1:3432.36,3355.57(νN-H);3059.23(ν=C-H);2934.32(νC-H);1612.32,1507.94(νC=C);1276.75,1217.40(νC-O);941.92,739.95(δ=C-H)。
1.3.4 1,2-二(2-氨基苯氧基)乙烷-N,N,N',N'-四乙酸甲酯(化合物4)的合成 将化合物3(2.44g,0.01mol)、二异丙基乙胺(6mL, 0.05mol)与溴乙酸甲酯(3mL,0.05mol)均匀混合于乙腈(10mL)中,80℃反应12h。待反应结束、冷却,向其中加入20mL 乙酸乙酯,抽滤、旋干、得到黄色油状物,加入少量甲醇、搅拌6h,抽滤、甲醇重结晶,得到白色固体4,产率87%,m.p.94~95℃。1H NMR(CDCl3,400MHz),δ:6.82~6.89(m, 8H, Ar-H),4.27(s, 4H, -OCH2CH2O-),4.15(s,8H, 4×-CH2-),3.56(s, 12H, 4×-CH3)。IR(KBr),ν,cm-1:3067.78(ν=C-H);2993.00,2951.29,2921.02,2888.20(νC-H);1748.17(νC=O);1596.85,1509.53(νC=C);1173.12(νC-O);742.49,706.25(δ=C-H)。
1.3.5 1,2-二(2-氨基-5-甲酰基苯氧基)乙烷-N,N,N',N'-四乙酸甲酯(化合物6)的合成 5~10℃,将POCl3(9.6mL)于40~45min 内滴加至DMF(20mL)中,再将化合物4(5.32g, 0.01mol)的DMF 溶液滴加至上述POCl3/DMF 混合物中,75℃反应4h。待反应完全、冷却,加入100mL 冰水、抽滤,柱分离,V乙酸乙酯∶V正己烷=2∶1 为洗脱剂,得到黄色固体6,产率90%,m.p.145~146℃。1H NMR(CDCl3, 400 MHz),δ:9.80(s,2H,2×-CHO),7.37~7.40(m, 4H, Ar-H),6.76(d, J=8Hz, 2H,Ar-H),4.31(s, 4H, 2×-OCH2-),4.23(s, 8H, 4×-CH2COO-),3.58(s, 12H, 4×-CH3)。13C NMR(CDCl3,101MHz),δ:190.45,171.16,149.51,145.02,129.85,127.02,116.55,110.37,77.30,76.98,76.66,66.94,53.44,51.99。IR(KBr),ν,cm-1:2954.41(νC-H);1747.19(νC=O);1519.63(νC=C);1168.65(νC-O);786.81(δ=C-H)。ESI-MS,m/z:611.1848 [M+Na]+。
1.3.6 1,2-二(2-氨基-5-苯并咪唑基苯氧基)乙烷-N,N,N',N'-四乙酸甲酯(S15)的合成 将化合物6(0.59g, 1mmol)与邻苯二胺(0.22g, 2mmol)均匀混合于无水乙醇(10mL)中,78℃反应4h,待反应结束、冷却、旋干,柱分离,V乙酸乙酯∶V正己烷=2∶1 为洗脱剂,得到黄色固体S15,产率为53%,m. p. 204~206℃。1H NMR(400MHz, CDCl3),δ:7.98(dd, J=8, 4.3Hz,4H, Ar-H),7.58~7.61(m, 2H, Ar-H),7.26(t, J=4.4Hz, 2H, Ar-H),6.87(d, J=8.6Hz, 2H, Ar-H),6.45(d, J=9.6Hz, 4H, Ar-H),3.85(s, 2H),3.24(s, 8H),2.60(s,12H),2.10(s,4H)。ESI-MS,m/z:765.2876[M+H]+。
1.4 S16 及S17 的合成
1.4.1 2-[2-(2-氨基苯氧基)乙氧基]-4-甲酰基苯胺-N,N,N',N'-四乙酸甲酯(化合物5)的合成 5~10℃,向DMF(20mL)中缓慢滴加POCl3(2.4mL),于40~45min 内滴加完全。将所得的POCl3/DMF 混合物在室温下搅拌1~2h、混合均匀,再滴加至化合物4(5.32g, 0.01mol)的DMF(20mL)溶液中,75℃反应4h。待反应结束、冷却,加入100mL 冰水、抽滤,柱分离,V乙酸乙酯∶V正己烷=1∶1 为洗脱剂,得到白色固体5,产率85%,m.p.131~132℃。1H NMR(CDCl3,400MHz),δ:9.80(s,1H,-CHO),7.37~7.40(m,2H,Ar-H),6.81~6.90(m, 4H, Ar-H),6.76(d, J=8.3Hz, 1H, Ar-H),4.31~4.32(m, 2H, -CH2O-),4.26~4.27(m, 2H,-OCH2-),4.24(s, 4H, 2×-CH2-),4.15(s, 4H, 2×-CH2-),3.57(d, J=4.5Hz, 12H, 4×-CH3)。13C NMR(CDCl3,100MHz),δ:190.47,171.92,171.22,150.12,149.64,145.00,139.27,129.89,126.71,122.17,121.59,118.93,116.49,112.85,110.51,76.98,76.66,67.25,66.64,53.33,51.95,51.76。IR(KBr),ν,cm-1:3015.76(ν=C-H);2954.95,2928.97(νC-H);1746.40(νOC=O);1681.01(νHC=O);1 593.97,1509.15(νC=C);1245.85,1164.87(νC-O);747.14(δ=C-H)。ESI-MS,m/z:583.1894[M+Na]+。
1.4.2 S16 及S17 的合成 合成方法类似于S15,2-[2-(2-氨基苯氧基)乙氧基]-4-甲酰基苯胺-N,N,N',N'-四乙酸甲酯(化合物5)的用量为0.56g、1mmol,N-甲基邻苯二胺或4-硝基邻苯二胺的用量分别为0.12g、1mmol,0.16g、1mmol。V乙酸乙酯∶V正己烷=2∶1 为柱分离洗脱剂。
2-[2-(2-氨基苯氧基)乙氧基]-4-(N-甲基苯并咪唑基)苯胺-N,N,N',N'-四乙酸甲酯(S16):黄色固体,产率68%,m. p.122~123℃。1H NMR(400MHz,CDCl3),δ:7.92~7.94(m, 1H, Ar-H),7.46(d, J=7.0Hz,1H, Ar-H),7.37~7.39(m, 2H, Ar-H),7.37(dd, J=6.3,2.8Hz, 2H, Ar-H),6.83~6.92(m, 5H, Ar-H),4.41(s,3H,-CH3),4.24(s,6H,2×-CH3),4.15(s,6H,2×-CH3),3.92(s, 4H, 2×-CH2-),3.58(s, 4H, 2×-CH2-),2.07(s,2H,-OCH2-),2.04(s,2H,-OCH2-)。IR(KBr),ν,cm-1:3393.08(νN-H);2951.93,2924.81(νC-H);1745.82(νC=O);1603.47,1508.99,1455.33(νC=C, νN=H);1172.28(νC-O);741.63(δ=C-H)。ESI-MS,m/z:663.2664[M+H]+。
2-[2-(2-氨基苯氧基)乙氧基]-4-(4-硝基苯并咪唑基)苯胺-N,N,N',N'-四乙酸甲酯(S17):红色固体,产率70%,m. p.165~167℃。1H NMR(400MHz,CDCl3),δ:8.49(s, 1H, Ar-H),8.20(dd, J=8.9, 2.0Hz,1H, Ar-H),7.98(s, 2H, Ar-H),7.76(s, 1H, Ar-H),7.68(t, J=8.6Hz, 2H, Ar-H),6.84~6.88(m, 3H, Ar-H),4.41(s, 1H, -NH-),4.25(s, 4H, 2×-CH2-),4.13(s, 4H, 2×-CH2-),2.99(s, 6H, 2×-CH3),2.86(s, 6H,2×-CH3),2.01(s, 2H, -OCH2-),1.99(s, 2H, -OCH2-)。IR(KBr),ν,cm-1:3374.15(νN-H);2999.98,2950.10(νC-H);1744.32(νC=O);1603.59,1507.13(νC=C);1171.49(νC-O);742.12(δ=C-H)。ESI-MS,m/z:716.2172[M+Na]+。
1.5 结构表征
合成中间产物经IR 与1H NMR 表征,均与文献相符。另由荧光材料S15、S16、S17 的1H NMR、IR 与MS 等表征数据可知,其结构正确,经合成引入的官能团羰基、醚键等均呈现其特征吸收,特征氢位移均能由1H NMR 进行归属,符合实验预期的结构设计,可进行进一步的光学性质研究。
IR 采用溴化钾压片法对各步合成产物进行IR 测试,波数范围4000~400cm-1。
1H NMR 以四甲基硅为内标、CDCl3为溶剂,对各步合成产物进行400MHz 的氢核磁及100MHz的碳核磁共振分析。
荧光分析 出射与入射狭缝宽度均为1nm,波长范围300~550nm。
2 结果与讨论
2.1 荧光材料S15、S16、S17 的荧光光谱
配制浓度为1×10-5mol·L-1的S15、S16、S17 水溶液,进行300~550nm 波段荧光光谱扫描,荧光材料S16、S17 的荧光光谱见图3。

图3 荧光材料S16、S17 的荧光光谱Fig.3 Fluorescent spectra of fluorescent materials S16 and S17
由图3 可知,因荧光发色团电子云密度的差异,分子中设计引入给电子基团-CH3的S16,相较于引入吸电子基团-NO2的S17,荧光谱峰显著红移[9,10]。测定显示,S16 的λmax=416nm,而引入-NO2,致使荧光发色团中电子云密度较小的S17,其λmax=368nm,荧光强度亦相对较弱。
图4 为荧光材料S15、S16 的荧光光谱。

图4 荧光材料S15、S16 的荧光光谱Fig.4 Fluorescent spectra of fluorescent materials S15 and S16
由图4 可知,具有双发色团结构的荧光材料S15(λmax=397nm),相比单发色团结构的S16 分子,荧光强度几乎达到2 倍增幅。由此可见,双发色团的结构修饰对荧光材料光学性能影响显著。
2.2 苯并咪唑缩合反应的反应机理
图5 为苯并咪唑缩合反应的反应机理。
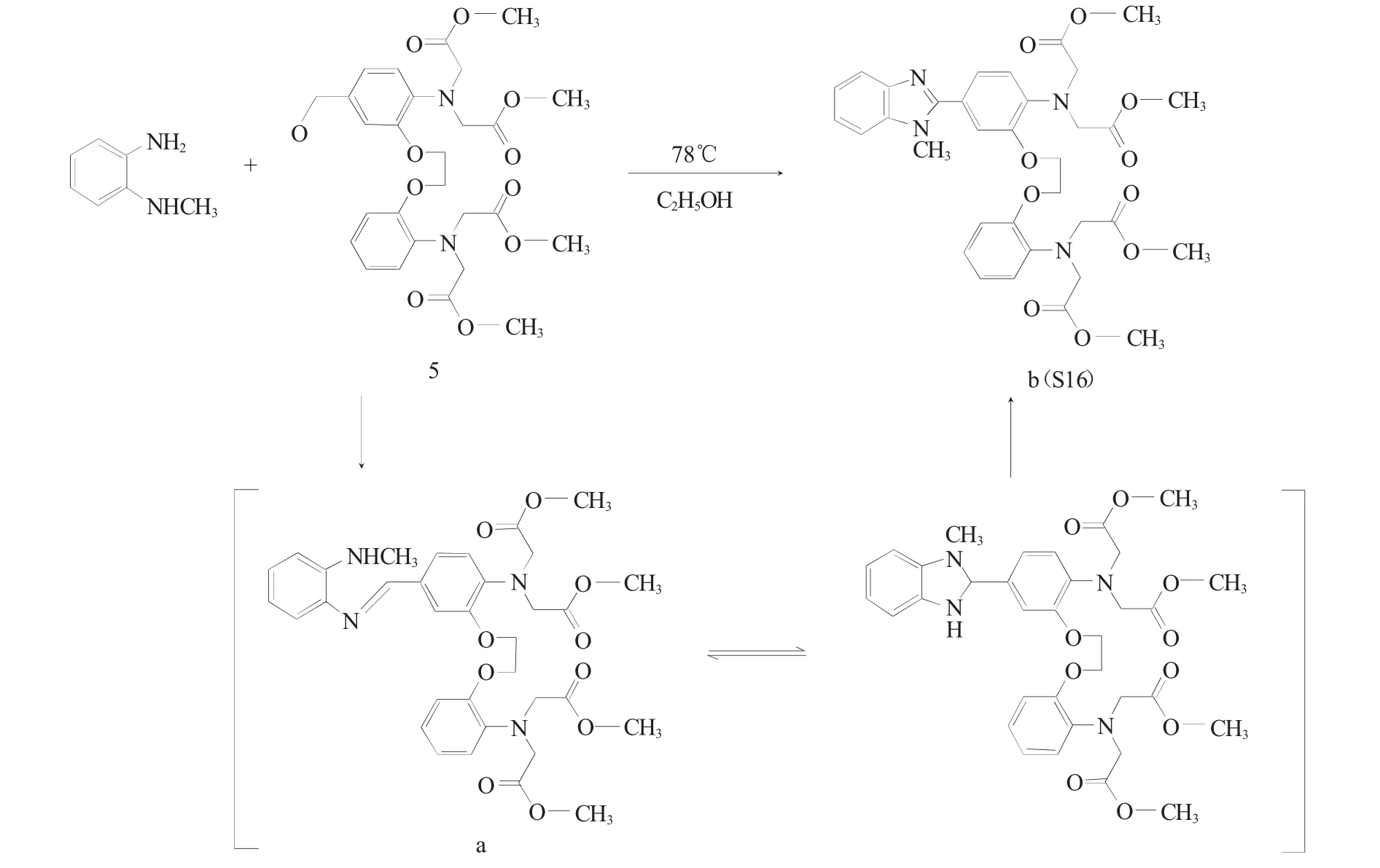
图5 苯并咪唑缩合反应的反应机理Fig.5 Reaction mechanism of the benzimidazole condensation
S15、S16、S17 合成路径中的苯并咪唑缩合反应,以取代邻苯二胺与芳香醛(S16、S17 制备路径中的化合物5,或S15 制备路径中的化合物6)为反应物。经反应温度考察,显示低温冰浴条件下进行该缩合反应时,反应进行4h 时,产物为油状物(a),而放置数天后,油状物发生性状的明显改变,得到新的黄色固体产物(b,即S16)。进一步分析化合物(a)、(b)的紫外-可见与红外光谱,谱图均显示特征谱带的差异。其中,紫外-可见光谱中,化合物(a)的吸收谱带峰值分别位于307.0、294.5、248.0 以及212.0nm,而(b)的吸收谱带峰值为291.0、245.0 和212.0nm。红外光谱中,(a)的特征吸收谱带为3066.8cm-1(ν=C-H)、1681.8cm-1(νN=H)、1030.1cm-1(νC-O-C),而(b)的氮氢伸缩振动处于1455.3cm-1(νN=H),较化合物(a)红移227cm-1。综合光谱分析结果,推测中间产物(a)为席夫碱,(b)为缩合反应产物苯并咪唑(图5),化合物(b)红外光谱特征氮氢伸缩振动νN=H的红移,可能是由于咪唑环上的碳氮键介于碳氮单键与碳氮双键之间,相比碳氮单键有所红移。此外,参考文献报道[11],苯并咪唑缩合产物受反应热力学与动力学因素影响较大[12,13],席夫碱产物(a)是速率产物与热力学产物,在反应中先生成,低温条件亦可稳定存在;苯并咪唑缩合产物(b)较(a)席夫碱产物稳定,经反应温度升高或反应时间延长而得到。反应发生时,芳香醛与邻苯二胺衍生物先加成后消去、给出席夫碱产物,继而发生关环、得到苯并咪唑缩合产物。为得到目标产物(b),经反应温度与溶剂选择,最终确定该缩合反应的最优反应条件为无水乙醇反应体系、78℃下进行回流反应,反应时长4h。
3 结论
通过缩合反应,将经典的BAPTA 受体与苯并咪唑类荧光发色团相连接,同时,设计引入吸电子基团-NO2、给电子基团-CH3,设计引入双发色团结构,借以调控苯环电子云密度与材料荧光强度,调控BAPTA 受体与目标物的结合能力。基于上述的差异性结构构筑,合成得到3 种荧光材料S15、S16、S17。光谱研究显示,吸电子基团-NO2与给电子基团-CH3对S16、S17 的荧光性能产生了一定影响,给电子基团-CH3的引入使S16 结构中苯环电子云密度增大,使其紫外-可见与荧光光谱相较于S17,出现最大吸收与发射波长向长波方向的红移。此外,分子中引入双发色团结构的荧光材料S15,荧光强度明显优于仅具有单发色团S16、S17,符合预期结构设计。这种差异性结构构筑的合成设计,实现了对荧光材料光学性能的有效调控,能够为新型光学材料的开发提供一定研究基础。
