太阳能电池钙钛矿材料模拟计算研究①
2022-05-12李达耀金成国黄伟新
李达耀, 金成国, 黄伟新
(1.北部湾大学,广西 钦州 535011; 2.广西科技大学,广西 柳州 545000)
太阳能具有无污染、分布广泛、清洁等优点。 目前,较为常见的太阳能电池材料有无机硅金属、钙钛矿等。 钙钛矿材料的结构式为ABX3,其中A、B、X分别为有机阳离子、金属离子、卤素基团。 钙钛矿材料具有促进太阳能利用的物理和化学特性,比如吸光性、电催化性等[1]。 钙钛矿太阳能电池有望引领未来太阳能电池的发展方向[2]。 但钙钛矿太阳能电池存在稳定性差、电池效率低等缺点,采用全无机钙钛矿掺杂结构是提高钙钛矿太阳电池材料性能的重要途径。 本文从以下2个方面探讨了不同类型掺杂对全无机钙钛矿光电性能和热稳定性的影响:①卤素掺杂结构对能带和结构稳定性的影响;②B 位掺杂结构的光电性能分析,及其对材料缺陷形成的影响。 在此基础上采用计算机模拟计算为合成具有高稳定性的晶体结构和高光电转换效率的新型钙钛矿材料提供理论指导。
1 模拟计算理论
模拟计算相比传统实验而言,具有效率、准确度高等巨大优势。 首先,模拟计算结果可以通过模型展示使研究人员对材料特性、材料各组分相关作用和材料在化学反应中的原理等有更好认识;其次,通过模拟计算可以减少实验次数、节省时间,降低科研成本。
在模拟计算过程中采用第一性原理方法,只要知道普朗克常量、电荷量、光速、玻尔兹曼常数、电子质量就可以应用量子力学原理计算出体系的性质。 但由于计算机本身计算能力有限,模拟计算也有一个缺点,即对计算量的要求不能太高。 因此,计算时选用的材料对应的原子数目应相对较小。 BO 近似以及密度泛函理论等在电子与电子相互作用的相关效应和交换计算中十分常见。
1.1 BO 近似
处理分子或其他系统时考虑其在量子力学中的条件,系统的波函数需要通过解薛定谔方程或其他类似的偏微分方程来获取。 根据Born-Oppenheimer 近似,考虑到电子与原子核之间质量存在巨大差距(通常原子核质量比电子大3 ~4个数量级)[3],电子的哈密顿量可根据BO 近似推断为:H=T+U+V,其中T为动能,V为外场影响,U为库伦作用。
1.2 密度泛函理论
密度泛函理论主要用于电子结构的研究,是计算钙钛矿材料电子结构必不可少的应用理论之一[4]。通常复杂多电子波函数都以常见的电子结构理论方法计算。 将复杂问题简单化,是密度泛函理论的主要目的。 电子密度相对容易计算,而波函数计算则恰恰相反,用前者来代替后者,问题会变得简单许多。 Kohn-Sham 方法是常见的密度计算方法,它可以将体系外部静电势中的电子相互作用引起的多体问题转化为电子运动问题。 而对于其中交换相关性,现阶段还没有适当方法来解决与交换有关的能量问题[5]。 在非特定条件下,与量子力学中解决问题的方法相比,密度泛函理论给出的结果十分理想,并且通过固态计算可以有效降低计算成本[6]。
2 计算方法
Material Studio 软件可以很好地进行原子替换、掺杂等应用操作,是一款十分实用的软件[7-8],无论是结构优化、性能测试,还是材料掺杂和量子力学计算,其数据都较为准确。
2.1 CASTEP 计算方法
在CASTEP 计算中,总能量的计算尤为关键[9-10]。CASTEP 程序在密度泛函理论的基础上通过计算得出的总能量分为3个部分:

式中ρ表示粒子的密度;T[ρ]表示一组密度为ρ而又没有相互作用的粒子的动能;U[ρ]是根据库仑相互作用得到的静电能;Exc[ρ]包含了所有多体系统对总能量的贡献[11]。
2.2 自洽计算
在CASTEP 计算中,平面波基函数在所有计算所需函数里是较为常见的。 由于哈密顿矩阵算符H还需要借助波函数C帮助完成运算,并不是独立的,平面波基函数可作为自洽计算(SCF)的求解方程[12-13]。
3 钙钛矿材料计算
钙钛矿材料的结构式为ABX3,目前很有应用前景的无机钙钛矿材料为CsPbI3,本文对CsPbI3进行模拟计算。
3.1 钙钛矿材料CsPbI3 模型
通过参数设置、系统模拟,构建钙钛矿材料CsPbI3模型。 首先将各项晶格参数填好,之后根据需求将原子添加进去,初步完成CsPbI3的模型构建。 进行参数设置后,构建的CsPbI3模型如图1 所示。 构建完成CsPbI3基础模型后,对钙钛矿结构进行掺杂、原子清除或原子取代等计算,基于CsPbI3基础模型,通过计算软件构建新的模型。

图1 CsPbI3 模型
3.2 钙钛矿材料计算参数
本文进行的是钙钛矿同质异构结构的计算,所以只以CsPbI3为例进行计算设置,后续的同质异构结构的计算都以之前设置的参数为基准。
3.2.1 计算参数
对于一个物理体系,能量分布决定了体系的状态或材料的性能。 态密度实际上也就对应着能量分布,有了态密度,就可以根据统计力学得到想要的热力学量。
构建完成CsPbI3结构模型后,用CASTEP 进行计算设置。 电荷密度由截止能决定,在计算中直接影响平面波个数及平面波的截止能。 在此次运算过程中,500 eV 作为一个判定能量值,代表截断半径的截止能,在500 eV 外的波函数变化与实际情况是一致的。

频率在(ω,ω+dω)的模数可以通过其色散关系得出,结合态密度g(ω)的定义,可得:
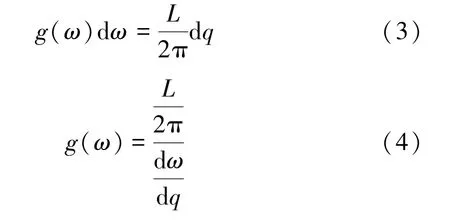
3.2.2 结构优化
在计算同质异构材料时,需要先对CsPbI3结构进行优化,当模拟CsPbI3结构中X 位的定量掺杂即X=F,Cl,Br,I的计算时,进行原子取代时可以在已有结构基础上进行,这样会大大降低工作量。
4 钙钛矿材料计算结果及分析
4.1 CsPbI3 同质异构体计算与结构分析
通过对不同结构的钙钛矿禁带宽度和系统结合能进行对比,从而推断不同组分钙钛矿材料的结构性能,进一步设计和优化其结构,提高光电转化效率。
3种不同晶体结构CsPbI3如图2~4 所示。

图2 CsPbI3 单斜晶系结构

图3 CsPbI3 斜方晶系结构

图4 CsPbI3 立方晶系结构
表1 为各晶系晶格结构参数和能量参数。 由图2~4及表1可以看出,不同晶系的晶胞大小不同,CsPbI3晶胞由大到小依次为立方晶系、单斜晶系、斜方晶系;单斜晶系的结合能最大,立方晶系的结合能最小,在晶系对称性上,立方晶系对称轴多,对称性好[15]。
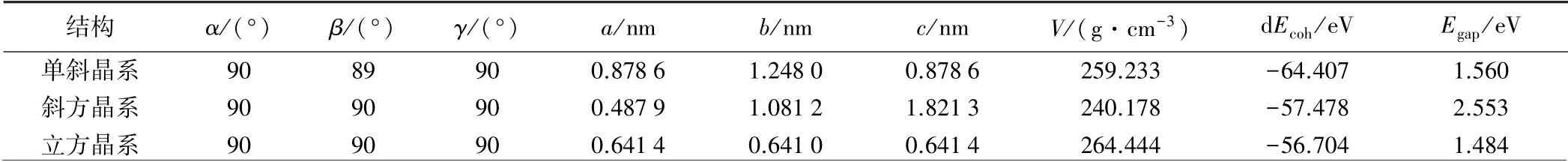
表1 CsPbI3 晶格的结构参数和能量参数
通过光子能量公式(式(5))以及物质波动量与其波长成反比关系(式(6)),可以根据带隙宽度求得发光颜色。

式中h为普朗克常量;ν为光子频率。
将式(6)代入式(5),可得:

通过式(7)计算得到CsPbI3单斜晶系、斜方晶系、立方晶系的波长分别为794.9 nm,492.1 nm,824.5 nm,可知斜方晶系带隙的发光颜色为绿光,可作发光材料,而单斜晶系和立方晶系则适合做光电材料。
表2 为不同晶体结构中Pb 与I、Cs 与I 原子间距离。 由表2可知,Cs 和I 原子间距离最小的为单斜晶系;而Pb 和I 原子间距离最小的是立方晶系结构,并且单斜晶系与斜方晶系并没有很大差异。
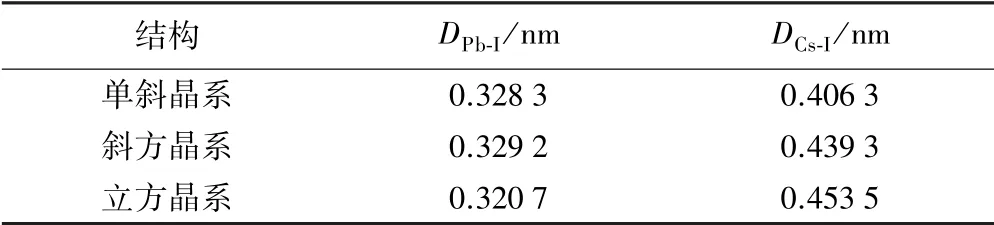
表2 CsPbI3 不同晶体结构中Pb 与I、Cs 与I 的原子间距离
态密度是指在一定能量范围内其轨道(能级数)的数量。 对CsPbI3材料做能带结构及态密度分析,软件模拟下的CsPbI3材料不同晶体结构下的能带间隙见图5~7。 能带间隙也称为带隙、能隙、禁带宽度,是指一个能带宽度,单位为eV。 由图5 ~7可见,引入杂质后能隙会变小,但态密度没有变化,这是因为掺杂原子质量太少,对电子的整体性质没有很大影响。 对于整个体系而言,在能带结构中引入了新能带,但这个新能带并不会引起决定性改变。

图5 CsPbI3 单斜晶系的能带结构

图6 CsPbI3 斜方晶系的能带结构

图7 CsPbI3 立方晶系的能带结构
4.2 X 位卤素掺杂
对CsPbI3的X 位即I 用F、Cl、Br 掺杂,晶格能量参数计算结果如表3 所示。 从表3可以看出,禁带宽度与系统结合能的绝对值存在正相关关系,禁带宽度越大,其系统结合能的绝对值就越大。 在对CsPbI3的X 位掺杂时,替换F、Cl、Br 原子的结构能带间隙越来越小,这说明掺杂原子半径越小,材料禁带宽度越大,系统结合能绝对值也越大。 其中,F 掺杂能大幅度提高带隙,CsPbI3-F 带隙值为2.659 eV,在太阳能光伏电板荧光材料选取中可以优先考虑;Br 掺杂CsPbI3-Br 带隙值为1.749 eV,比较适合作为光伏电板光电材料。

表3 CsPbI3 的X 位掺杂时晶格能量参数
4.3 B 位掺杂
对CsPbI3的B 位即Pb 用Ca、Yb、Ba、Sn、Sr 掺杂,晶格参数计算结果如表4 所示。 根据表4可知,B 位掺杂对调节结合能具有作用,其中Ca 掺杂时结合能降低最为明显。 B 位掺杂对带隙有明显调节作用,其中Sn 掺杂可使禁带宽度降至1.184 eV。 受计算机限制,计算采用的超晶格模型较小,特别是对Yb 的掺杂,计算结果需要与实验进一步对照。

表4 CsPbI3 的B 位掺杂时晶格的结构参数和能量参数
5 结 论
采用第一性原理计算和CASTEP 计算程序研究了CsPbI3的3种同质异构体和B 位、X 位掺杂对材料结构稳定性和能带结构的影响,结果表明:
1) 根据模拟计算数据,3种同质异构体的结构稳定性依次为单斜结构、斜方结构、立方相结构。 CsPbI3单斜晶系、斜方晶系、立方晶系的波长分别为794.9 nm、492.1 nm、824.5 nm,斜方晶系带隙的发光颜色为绿光,可作绿色光的发光材料,单斜晶系和立方晶系则适合作太阳能光伏电板的光电材料。
2) 对CsPbI3的X 位掺杂时, F 掺杂能大幅度提高带隙,CsPbI3-F 的带隙值为2.659 eV,在太阳能光伏电板荧光材料选取中可以优先考虑;Br 掺杂调节带隙到1.7 eV,CsPbI3-Br 比较适合用作光伏电板光电材料。
3) B 位掺杂可以有效提高立方相稳定性,在实践中可以通过B 位掺杂提高CsPbI3稳定性。 B 位掺杂对禁带宽度有明显调节作用,其中Sn 掺杂可使禁带宽度降至1.184 eV;B 位掺杂对调节结合能有明显作用,其中Ca 掺杂结构结合能降低最明显。
