柯里拉京对照品的制备及其结构鉴定
2022-03-30郑有德陈小红莫太刚樊三虎骆学庆孟伦秀张定堃成都三勒浆药业集团四川华美制药有限公司成都60045乳品营养与功能四川省重点实验室成都6002成都三勒浆药业集团成都中医药大学产学研联合实验室成都60045
郑有德,陈小红,莫太刚,2,,樊三虎,2,,骆学庆,孟伦秀,张定堃(. 成都三勒浆药业集团四川华美制药有限公司,成都 60045;2. 乳品营养与功能四川省重点实验室,成都 6002;. 成都三勒浆药业集团-成都中医药大学产学研联合实验室,成都 60045)
柯里拉京(corilagin,分子式:CHO),又称柯子次鞣素、诃里拉京,属天然植物多酚单宁酸类化合物,为类白色固体粉末,可溶于甲醇、乙醇、二甲基亚砜等有机溶剂。柯里拉京是一种可以从许多植物提取的水溶性逆没食子酸鞣质,如蜜柑草属植物、牦牛儿苗属植物、老鹳草属植物、大戟属植物、铁苋属植物、安石榴属植物、槭属植物及榄仁树属植物等。柯里拉京为叶下珠、老鹳草、蜜柑草、白三草等植物的有效成分,也存在于余甘子、龙眼、橄榄、诃子中等。研究表明柯里拉京具有多种生物活性,如抗肿瘤、抗氧化、抗动脉粥样硬化、抗纤溶、降血压、抑制病毒、抗菌、抗炎等。
本文以叶下珠全草为研究对象制备高纯度柯里拉京对照品,现报道如下。
1 材料
大戟科油柑属植物叶下珠(Phyllanthus urinaria
L.)干燥全草购于成都五块石药材市场,产地四川广元,经吴燕成总工程师确认。95%乙醇(批号:20021018)、盐酸(批号:20020816)(分析纯,成都光华化学试剂厂),乙腈(分析纯,上海陆都实业有限公司,批号:20021028),丙酮(分析纯,重庆东试化工有限公司,批号:20021116),D101大孔树脂(天津南开大学精细化学试验厂,批号20021206),Sephadex LH-20(瑞典,批号:292931),Diaion HP-20(日本三菱化学公司,批号:2H508),硅胶GF254(化学纯,国家药典委员会监制青岛海洋化工集团公司,批号:9639);紫外灯、Water 2996高压液相色谱仪(Waters)。2 方法与结果
2.1 提取分离与纯化
① 称取叶下珠干燥全草300 g,铡成3~5 cm的段,加入20倍以上纯水浸泡0.5 h,分别微沸煎煮1.5、1 h,滤过,合并两次滤液,减压浓缩至约1.2 kg,离心除去沉淀。② 浓缩液通过D101大孔吸附树脂,用至少20 L纯化水洗涤,检测后弃去水液。用70%乙醇洗脱至无柯里拉京检出,洗脱液减压浓缩,置入蒸发皿中挥去残留乙醇,得浓缩液200~400 mL。③ 浓缩液放冷,滤过,滤液通过Diaion HP-20 大孔吸附树脂,用10 L以上纯化水洗涤,检测后弃去水溶液。用70%乙醇洗脱,至无柯里拉京洗出,洗脱液减压浓缩,入蒸发皿中挥去残留乙醇,浓缩液放冷加入7倍乙腈搅拌均匀,沉淀0.5 h,滤除沉淀,滤液减压浓缩回收乙腈,再入蒸发皿中挥去残留乙腈,得浓缩液约100 mL。④ 浓缩液放至室温,小心置Sephadex LH-20柱上端,用约10 L蒸馏水洗涤,检测后弃去洗涤液。用20%丙酮液洗脱,至无柯里拉京检出,洗脱液减压浓缩,入蒸发皿中挥去残留丙酮至有较多晶体析出,冷至室温,充分冷藏,抽滤出结晶。⑤ 柯里拉京粗结晶,用50%~60%乙腈反复重结晶至得到近白色结晶。结晶挥去溶剂后放入五氧化二磷干燥器干燥至恒重,得柯里拉京2.4 g,平均收率约0.8%。
2.2 纯度分析
2.2.1 薄层色谱 取柯里拉京,加甲醇制成每mL含5 mg的溶液,照薄层色谱法(《中国药典》2020年版四部附录)试验,取此溶液4、8、12、16、20 μL,点于硅胶GF薄层板或十八烷基键合硅胶(PR-18 FS)薄层板上,分别以乙腈-10%醋酸(9∶1)或乙腈-HO(2∶8)为展开剂,展开,取出,晾干,于紫外光(365 nm)下观察,或喷以1%FeCl-乙醇溶液,结果薄层板显单一色斑,除主斑点外均未见杂质斑点。
2.2.2 纯度测定色谱柱:Lunar C(5 μm,4.6 mm×150 mm);流动相:甲醇-0.1%冰醋酸(20∶80),检测波长:220 nm、254 nm、272 nm、330 nm;柱温:30℃,进样量:10 μL。结果均未见明显杂质峰。
2.2.3 含量测定 照高效液相色谱法(《中国药典》2020年版四部)测定。Water 2996型高效液相色谱仪及Water 2996可变波长检测器,Empower工作站,手动进样器,C色谱柱(Lunar,Phenomenex,150 mm×4.6 mm,5 μm),溶剂均为色谱纯,试剂均为分析纯。色谱条件为流动相:乙腈-0.5%醋酸(1∶9),检测波长:272 nm,流速:1 mL·min,柱温:25℃。取柯里拉京约0.8 mg,各3份,精密称定,加水溶解并稀释至10 mL,摇匀,即得。测得柯里拉京峰面积分别为2756 356、2724 593、2745 250,RSD
为0.59%,按归一法计算含量100%。2.3 结构鉴定
mp 210℃(分解);:-245(C,1.5,EtOH);IR(KBr)cm:3416(羟基),1715,1695(羰基),1616,1532(苯环)等;MS(FAB)[M-H]=633;MS(ESI)[M-H]=633.07;H-NMR(400 MHz,MeOD)δ
:7.08(2H,s,Galloy,A,H-2,6),6.72(1H,s,HHDP,B,H-2),6.69(1H,s,HHDP,C,H-2),6.39(1H,d,J
=4.0 Hz,Glucose-H-1),4.96~5.02(2H,m),4.55~4.57(1H,m),4.49(1H,d,J
=4 Hz),4.17~4.21(1H,m),4.01~4.03(1H,m);C-NMR(100 MHz,MeOD)δ
:95.0(glc-C-1),71.5(glc-C-2),76.1(glc-C-3),65.0(glc-C-4),76.1(glc-C-5),62.4(glc-C-6),120.5(Gal-C-1),110.9(Gal-C-2,6),146.3(Gal-C-3,5),140.4(Gal-C-4),166.6(Gal-C-7),125.4(HHDP,B,C-1),125.5(HHDP,C,C-1'),108.3(HHDP,B,C-2),110.1(HHDP,C,C-2),143.5(HHDP,B,C-3),143.8(HHDP,C,C-3'),135.1(HHDP,B,C-4;C,C-4'),144.3(HHDP,B,C-5;C,C-5'),115.0(HHDP,B,C-6),115.3(HHDP,C,C-6'),168.5(HHDP,B,C-7),170.1(HHDP,B,C-7')。柯里拉京为白色结晶状固体,mp 210℃(分解),: -245(C,1.5,EtOH),其溶液遇三氯化铁呈深兰色,紫外吸收光谱λ
(MeOH)219 nm、272 nm;IR(KBr)cm:提示有羟基(3417),羰基(1715,1695),苯环(3077,1616,1532);MS(FAB)示M=633(M-1),结合H-NMR、C-NMR及DEPT谱,推定其分子式为CHO,分子不饱和度为17。重水交换试验提示具有11个羟基,其中9个在H-NMR谱中出现在δ
8.16~9.34的低场位置,提示为酚羟基。根据C-NMR中的δ
92.2及其余δ
62.2~77.7等5个碳的化学位移,可判断分子中具有吡喃葡萄糖的结构部分;根据在H-NMR及H-H-COSY谱δ
7.03(2H,s)、6.56(1H,s)及6.49(1H,s)出现4个不相关的芳质子信号,可以判断该化合物分子中有没食子酰基(galloyl)及HHDP(六羟基苯甲酰基)基团的取代。由于H-NMR中除糖H-1及糖H-2的偶合常数J
外,其余糖上连位质子2,3,4,5间的偶合常数均较小这一特点,表明糖为C构象为船式构象,提示HHDP取代于糖的C-2及C-4或C-3及C-6位。H-C 化学位移相关试验结果可见:糖H-1质子(δ
6.20)与δ
164.8的酯羰基相关,后者又与没食子酰基(没食子酰,A)上的芳质子(δ
7.03)相关,证明没食子酰基取代于糖C-1位;同样,糖H-3质子(δ
4.59)与δ
166.8的酯羰基相关,后者又与HHDP(B)上的芳质子(δ
6.58)相关;糖H-6质子(δ
4.24)与δ
167.1的酯羰基相关,后者又与HHDP(C)上的芳质子(δ
6.52)相关,证明HHDP基取代于糖的C-3及C-6位。根据以上理化常数及光谱数据,可确定本化合物为鞣质类化合物柯里拉京(结构式见图1)。
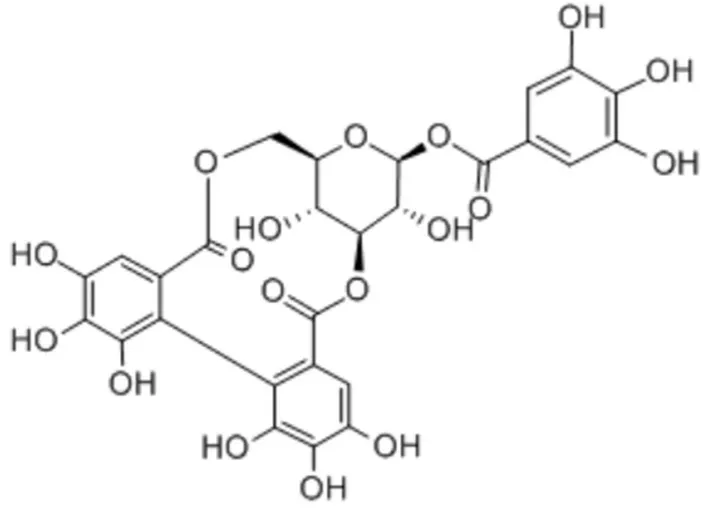
图1 柯里拉京的结构式Fig 1 Structure of corilagin
3 小结
叶下珠是提取柯里拉京较好的原料,分离难度小,收率稳定。文献报道陕西、四川、广西产的叶下珠中柯里拉京含量最高。本研究在纯化的同时也对诃子、余甘子药材中柯里拉京的成分进行了初步研究,结果提示以诃子提取物喷干粉及诃子提取液制备柯里拉京时发现其对葡聚糖凝胶污染较大,且分离难度大故而收率较低。余甘子及余甘子汁也尚难以作为提取柯里拉京的原料。
葡聚糖凝胶(Sephadex LH-20)是鞣质分离常用的树脂,其缺点是对某些鞣质吸附太强而不易洗脱,从而影响凝胶寿命。精制纯化过程中分别选用乙醇、甲醇和乙酸、丙酮作为洗脱剂,发现20%丙酮作为洗脱剂时效果较好。此外,当重结晶溶剂乙腈浓度为80%时会使母液浓缩后析出结晶过快,经摸索发现50%~60%乙腈较适宜。
致谢:
四川省中药所罗泽渊、周德庆、罗学成;四川华美制药有限公司总工程师吴燕成。本研究中结构鉴定由中国食品药品检定研究院分部等单位协助完成。