N-甲基-D-天冬氨酸受体信号通路及其介导的细胞凋亡机制研究进展
2022-03-23韩宜晓侯雅竹闫海峰王贤良毛静远
韩宜晓,侯雅竹,闫海峰,王 帅,王贤良,毛静远
天津中医药大学第一附属医院心内科 国家中医针灸临床医学研究中心,天津 300381
N-甲基-D-天冬氨酸受体(N-methyl-D-aspartate receptor,NMDAR)是重要的离子型谷氨酸受体,具有配体及电压双重门控特性,组成及功能复杂,分布广泛,在多种疾病或应激状态的病理生理进程中起着关键作用。研究表明,其信号通路可通过线粒体及内质网损伤、活性氧(reactive oxygen species,ROS)与过氧亚硝酸盐(ONOO-)生成、丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)及钙蛋白酶(calpain)激活等介导细胞凋亡。本文就NMDAR的结构分布、生物特性以及其介导的信号通路在细胞凋亡中的作用机制等方面进行综述,以期为相关疾病的防治提供参考。
NMDAR结构分布及生物特性
结构NMDAR是由NMDAR1(GluN1)、NMDAR2(GluN2)、NMDAR3(GluN3)同源亚基构成的四聚体复合物,每个亚基又分别有多个剪接变体或亚型,主要包括GluN1- 1a/b~4a/b剪接变体、GluN2A~D及GluN3A~B亚型[1- 2]。GluN1亚基存在于所有功能性NMDAR复合物中,基因编码为谷氨酸受体离子化NMDA1受体基因(glutamate receptor ionotropic,N-methyl-D-aspartate 1,GRIN1),其1个N末端(外显子5)和2个C末端(外显子21和外显子22)可以进行选择性剪接,形成8个剪接变体,在NMDAR复合物中起通道作用[3];GluN2包括GRIN2A~D的4个基因编码,修饰调节NMDAR;GluN3可与GluN1/GluN2结合形成复合物,降低Ca2+通透性,负向调节NMDAR通道开放[4- 5]。
分布NMDAR广泛表达于神经元细胞、神经胶质细胞、人类角质形成细胞、淋巴细胞、血管内皮细胞、气道平滑肌细胞等细胞组织中,也存在于心脏、肺脏、脾脏、肾脏、胃、卵巢等器官中[6]。NMDAR的分布主要与亚基类型相关,其中,GluN1分布广泛,主要分布于心脏、肾近端小管基底侧、成骨细胞和破骨细胞等[7];GluN2A分布于颈动脉、肾小球、破骨祖细胞[8],GluN2B主要表达于肾皮质、新生儿心脏组织、破骨细胞、淋巴细胞[9],GluN2C分布于胰腺、骨骼肌、肾皮质与髓质,GluN2D存在于肾皮质、破骨细胞,成骨细胞中[8];GluN3分布较少,主要存在于肾脏[10]。
生物特性NMDAR分为细胞外配体结合域和跨膜离子通道两个部分,其激活需同时满足配体及电压双重门控特性,进而介导Ca2+、Na+内流和K+外流,且Ca2+的相对通透性为Na+的10倍[11- 12]。作为配体门控离子通道,NMDAR的激活需甘氨酸(或D-丝氨酸)及谷氨酸(或天冬氨酸)分别与GluN1及GluN2亚基相结合[13],当配体与NMDAR结合时,配体结合域如蛤壳般闭合,跨膜离子通道可能开放。作为电压门控离子通道,其可由Mg2+、Zn2+、H+等离子调节,具有Mg2+电压依赖性阻滞的特征,具体表现为在膜电位约为-70 mV时,NMDAR离子通道被Mg2+阻断,其激活需经膜去极化[14- 15]。NMDAR的激活,极大提高Ca2+的通透性,促使细胞膜进一步去极化及内流[16]。
NMDAR介导的细胞凋亡通路
生理条件下,NMDAR激活可促进细胞保护,防止细胞凋亡和兴奋性毒性损伤;病理条件下,NMDAR过度活化、被特殊信号或位点激活可触发细胞凋亡程序。NMDAR的细胞凋亡机制主要由其介导的Ca2+内流触发,进而引发一系列促凋亡级联反应(图1),以下分别进行论述。
NMDAR与线粒体及内质网通路线粒体及内质网功能障碍在细胞凋亡的信号级联反应中起关键作用。线粒体内膜通道中Ca2+单向转运通道是线粒体能量代谢与交换的中心环节,该通道可确保Ca2+沿电化学梯度从细胞质转运到线粒体基质中。内质网是细胞内的重要钙库,对维持钙信号的准确性起关键作用。病理状态下,NMDAR过度激活,引发大量Ca2+内流,导致线粒体基质中Ca2+超载,从而使线粒体通透性转换孔(mitochondrial permeability transition pore,mPTP)打开、膜通透性转变、线粒体肿胀并外膜破裂,产生大量ROS,激活凋亡生化级联[17- 19]。同时,大量Ca2+通过线粒体膜,破坏了质子梯度,引起膜电位下降甚至崩解,诱导线粒体功能障碍,释放细胞色素C、细胞凋亡诱导因子(apoptosis inducing factor,AIF)及半胱氨酸天冬氨酸蛋白酶(cysteinyl aspartate specific proteinase,Caspase)酶原等凋亡因子到胞质中,引起细胞凋亡[20- 21]。内质网中的大量Ca2+使网内钙稳态失衡,发生内质网应激,激活内质网胞质面的Caspase- 12,启动Caspase级联反应,进一步活化Caspase- 9 及Caspase- 3,完成对其相应底物的切割,导致细胞凋亡[17,22]。
此外,线粒体通过电位驱动的联合转运机制摄取过量Ca2+引起的线粒体功能障碍是细胞凋亡的关键事件。NMDAR过度激活引起的Ca2+会抑制线粒体三磷酸腺苷(adenosine triphosphate,ATP)的产生、ATP酶逆转、甚至胞内ATP耗竭,亦可引起细胞凋亡[23- 24]。
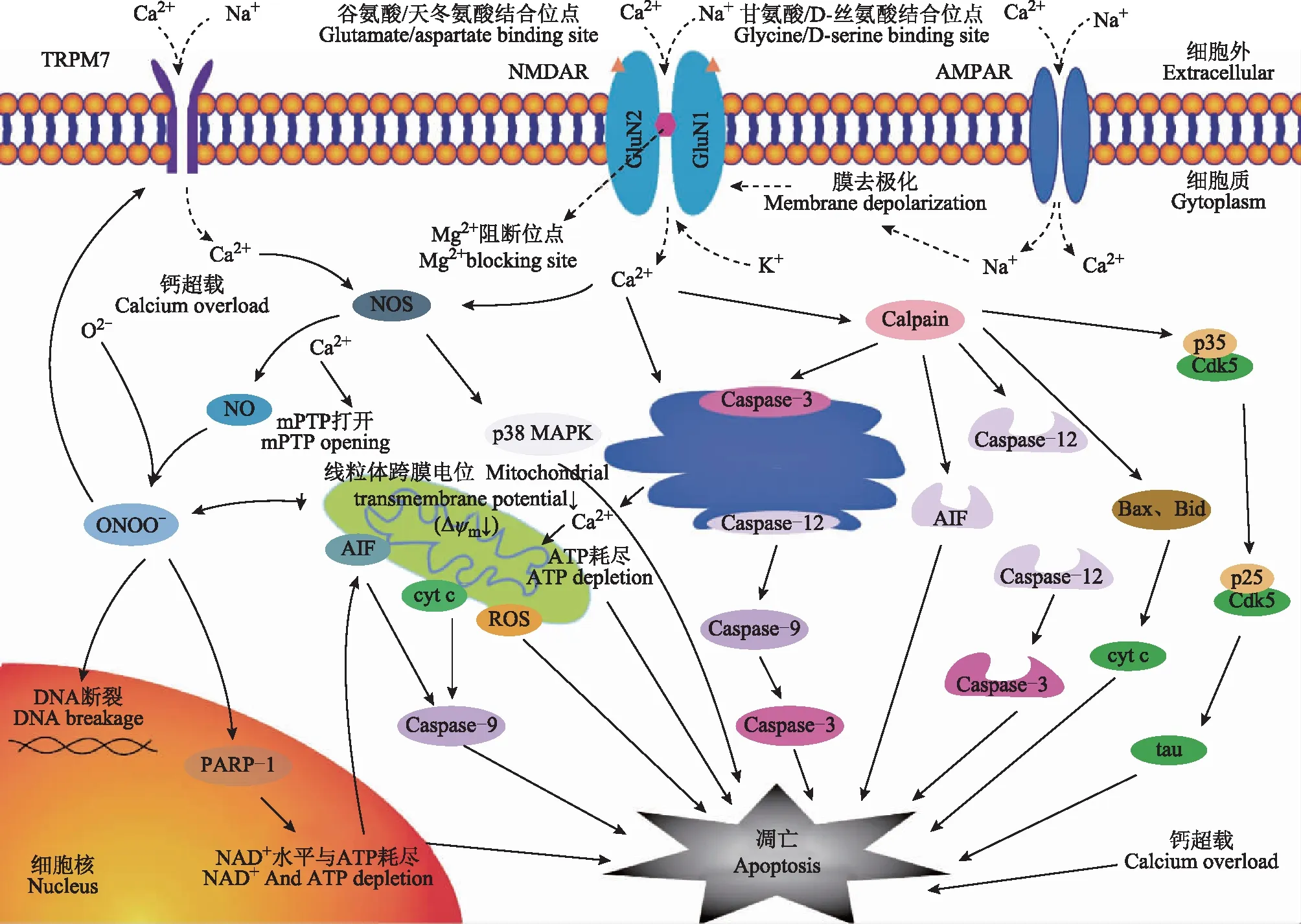
NMDAR:N-甲基-D-天冬氨酸受体;TRPM7:瞬时受体电位M7;AMPAR:α-氨基-3-羟基-5-甲基-4-异唑丙酸受体;NOS:一氧化氮合酶;NO:一氧化氮;p38 MAPK:p38丝裂原活化蛋白激酶;Caspase:半胱氨酸天冬氨酸蛋白酶;mPTP:线粒体通透性转换孔;ONOO-:过氧亚硝酸盐;ATP:三磷酸腺苷;DNA:脱氧核糖核酸;PARP- 1:聚腺苷二磷酸核糖聚合酶- 1;Cdk5:周期蛋白依赖性激酶5;AIF细胞凋亡诱导因子;Cyt c:细胞色素c;ROS:活性氧;NAD+:烟酰胺腺嘌呤二核苷酸
NMDAR与一氧化氮合成酶信号通路生理状态下Ca2+升高时一氧化氮合酶(nitric oxide synthase,NOS)可产生少量一氧化氮(nitric oxide,NO),而NMDAR活性调节可使Ca2+依赖的NOS过度激活,通过多条途径引发凋亡,具体包括:线粒体功能障碍、p38丝裂原活化蛋白激酶(p38 mitogen-activated protein kinase,p38 MAPK)信号激活、瞬时受体电位M7(transient receptor potential melastatin 7,TRPM7)激活等[25- 26]。NOS激活可导致NO及ROS的产生,过量的NO产生毒性作用,并与其他活性氧(如超氧化物)结合形成ONOO-,破坏细胞成分,抑制线粒体呼吸链酶,促进线粒体去极化[27];同时,ONOO-破坏DNA,导致单链断裂和聚腺苷二磷酸核糖聚合酶- 1[poly(ADP-ribose)polymerase- 1,PARP- 1]过度激活,耗尽细胞内烟酰胺腺嘌呤二核苷酸(nicotinamide adenine dinucleotide,NAD+)水平并触发线粒体释放AIF促进细胞凋亡[28]。
依赖于NMDAR的Ca2+内流通过NOS激活还可导致细胞内氧化应激,进而磷酸化p38 MAPK,激活2种相互关联的线粒体裂变蛋白:动力样蛋白1(dynamin-like protein 1,DLP1)和线粒体裂变因子(mitochondrial fission factor,MFF),使线粒体动力学异常,线粒体碎裂,促进线粒体自噬,下调抑凋亡蛋白Bcl- 2,激活凋亡蛋白Bax,致使Bcl- 2/Bax比值降低,促细胞凋亡;同时,p38 MAPK的激活也促进了NMDAR刺激后NO的产生,进而诱导细胞凋亡[29- 31]。
此外,NMDAR信号也会通过NOS激活TRPM7促凋亡靶点。TRPM7为瞬时受体电位TRP家族中一种包含阳离子通道和蛋白激酶双重结构的膜蛋白,广泛表达于哺乳动物的心脏、肝脏、脾脏、肺脏、肾脏、脑等多种器官和组织中,对Ca2+、Mg2+、Na+等二价和单价阳离子具有通透性[32]。依赖于NMDAR的Ca2+内流通过NOS激活引发NO产生并与超氧化物结合,形成TRPM7的活化剂ONOO-,同时Ca2+经TRPM7内流,又激活NOS,形成一个产生ONOO-和引起细胞内钙超载的正反馈循环,最终导致细胞凋亡[33]。
NMDAR与MAPK信号通路MAPK是真核细胞中的一类丝氨酸/苏氨酸激酶,包括细胞外信号调节激酶(extracellular signal-regulated kinase,ERK)、c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)、p38 MAPK亚族[34]。NMDAR激活ERK、JNK诱导细胞凋亡,并根据激酶分布情况发挥不同作用:心肌细胞NMDAR可使ERK磷酸化抑制细胞凋亡[35],而其驱动的Ca2+内流对JNK磷酸化并无显著影响[29];神经元突触中NMDAR激活ERK抑制细胞凋亡,突触外NMDAR抑制ERK诱导细胞凋亡[36],缺血性神经元中NMDAR可激活JNK的促凋亡反应[29]。而NMDAR激活p38 MAPK诱导的细胞凋亡则通过上述NOS信号通路介导[29- 31]。
NMDAR与calpain信号通路calpain是一类钙依赖的中性蛋白酶,在不同组织类型、生物体中广泛表达。NMDAR持续病理性激活导致的蛋白质、酶等大量水解,是细胞凋亡的重要因素[37]。细胞内Ca2+水平升高,并转移至细胞膜,与calpain结合,在磷脂存在时被激活。过度活化的calpain切割并激活凋亡蛋白Bax、Bid、内质网中Caspase- 3,并降解血影蛋白、钙依赖的转录因子、Caspase家族成员、Bcl- 2家族成员、AIF等底物蛋白,引起一系列细胞损伤[38- 39]。此外,NMDAR诱导的calpain激活还可导致细胞周期蛋白依赖性激酶5(cyclin-dependent kinase 5,Cdk5)的调节亚基p35裂解为p25,使Cdk5过度激活,进而磷酸化tau蛋白来诱导神经细胞凋亡[40]。
NMDAR介导与细胞凋亡相关的疾病
NMDAR信号通路介导的细胞凋亡与多种疾病的发生发展相关,具体包括:缺血性心脏病、缺血性脑损伤、高同型半胱氨酸血症(hyperhomocysteinemia,HHcy)、获得性免疫缺陷综合征(acquired immunodeficiency syndrome,AIDS)、神经退行性疾病、肾损伤、糖尿病-β细胞功能异常、视网膜损伤等。
缺血性心脏病NMDAR诱导的心肌细胞凋亡在缺血性心脏病中具有重要作用。心肌缺血发生时,ATP显著减少,谷氨酸、甘氨酸的转运及代谢出现障碍,在细胞膜外堆积,堆积的谷氨酸首先激活α-氨基- 3-羟基- 5-甲基- 4-异唑丙酸受体(α-amino- 3-hydroxy- 5-methyl- 4-isoxazole-propionicacid receptor,AMPAR),致Na+内流,细胞膜去极化,解除生理条件下Mg2+对NMDAR通道的阻断作用[41]。Liu等[42]研究提示离体缺血SD大鼠心脏中Ca2+水平在30 min后上升,且加入NMDA后缺血心肌Ca2+显著升高,提示NMDAR活性在不可逆心肌缺血损伤的情况下增强心肌细胞Ca2+内流。在体外培养氧葡萄糖剥夺(oxygen-glucose deprivation,OGD)心肌细胞模型中加入NMDA后发现,Ca2+及细胞凋亡水平显著增加,并激活p38 MAPK,验证了在心肌缺血条件下NMDAR病理性激活,Ca2+大量内流,进而磷酸化p38 MAPK导致心肌细胞凋亡[29]。
缺血性脑损伤作为兴奋性神经递质,大量谷氨酸过度激活NMDAR诱导神经细胞凋亡是缺血性脑损伤的主要机制之一[43]。缺血期间,有限脑血流量耗尽神经元维持离子稳态所需的氧气和营养物质,破坏离子梯度使膜去极化,致使兴奋性神经递质谷氨酸释放到突触间隙。同时,能量消耗会损害再摄取转运体功能,使其无法清除过量的谷氨酸,导致兴奋性谷氨酸在细胞外积累过度激活NMDAR,从而通过上述多种途径诱导细胞凋亡[44]。
Simon等[45]在缺血性脑损伤啮齿动物模型的海马体中,注入选择性NMDAR拮抗剂2-氨基-7-膦酰庚酸,30 min后大脑切片提示在药物阻断NMDAR传递的海马区,神经元凋亡较对侧海马区明显减弱,提出阻断NMDAR可防止缺血性脑损伤。Mishra等[46]将GluN2选择性抑制剂艾芬地尔腹腔注射进入缺血性脑损伤大鼠模型中,脑缺血区的丙二醛(malondialdehyde,MDA)水平显著降低,TUNEL染色阳性细胞数明显减少,进而提出NMDAR的GluN2阻断可防止缺血性脑损伤。Yu等[47]通过给予缺血性脑损伤大鼠新型NMDAR抑制剂BQ- 869,其Ca2+浓度显著下降,缺血梗死面积与细胞凋亡数量显著降低。上述研究均证实靶向抑制NMDAR可减轻缺血性脑损伤。
HHcy同型半胱氨酸(homocysteine,Hcy)属于一种谷氨酸受体亚型,其激活NMDAR介导的细胞凋亡[48]在HHcy进程中起到重要作用。
Moshal等[49]报道,通过GluN1基因敲除,可缓解HHcy导致的心肌细胞线粒体内ROS、NO及ONOO-水平的异常升高,减少细胞凋亡。同时,Tyagi等[50]研究结果显示,心脏特异性GluN1缺失,可改善HHcy导致的心肌细胞线粒体内亚硝化应激及线粒体基质金属蛋白酶- 9(mitochondrial matrix metalloproteinase- 9,mtMMP- 9)活性的异常升高,减轻了线粒体连接蛋白43(mitochondrial connexin- 43,mtCxn- 43)的易位和降解,减少细胞凋亡。两项研究均表明HHcy可通过激活GluN1来介导心肌细胞凋亡。近期Jindal等[51]研究发现HHcy可致脑缺血性损伤加重,并随时间延长而升高,而给予GluN2A抑制剂NVP-AAM077可显著减小缺血性梗死面积,提出GluN2A激活诱导神经元凋亡是HHcy条件下缺血性损伤严重程度的关键决定因素。
AIDSNMDAR介导的细胞凋亡可显著影响AIDS的病程发展。AIDS病毒中的人类免疫缺陷病毒1型(human immunodeficiency virus 1,HIV- 1)包膜糖蛋白gp120激活NMDAR是引起细胞凋亡的原因之一。Meng等[52]将含gp120的大鼠心肌细胞系H9c2经MK- 801预处理后,自噬相关蛋白7(autophagy related protein 7,ATG7)、自噬基因Beclin 1和自噬标志物溶酶体相关膜蛋白1(lysosomal associated membrane protein 1,LAMP1)均显著下降,提示HIV- 1 gp120可激活NMDAR,进而诱导细胞过度自噬,最终导致细胞凋亡。此外,HIV转录因子Tat(HIV-tat)可诱导神经元去极化,激活NMDAR,使大量Ca2+内流,导致胞内钙稳态破坏,进而通过NMDAR介导细胞凋亡的多条通路,亦可使神经元产生不可逆损伤[53- 54]。
神经退行性疾病NMDAR介导细胞凋亡参与多种神经退行性疾病进程,如阿尔茨海默病(Alzheimer’s disease,AD)、亨廷顿病(Huntington’s disease,HD)等。AD中β-淀粉样蛋白(amyloid beta,Aβ)寡聚体调节谷氨酸释放,寡聚肽Aβ在神经元中诱导NMDAR依赖性凋亡,此凋亡途径参与升级介导AD中Aβ毒性。AD中NMDAR过度激活,Ca2+大量涌入,线粒体Ca2+超载,进而引起功能障碍[55- 56]。亦有报道表明,过量NMDAR激活Calpain,使Cdk5的调节亚基p35裂解为p25,增加tau高磷酸化,促神经元凋亡,抑制NMDAR可防止AD神经元损伤[57- 60]。HD由亨廷顿蛋白(huntingtin,Htt)基因突变、纹状体中型多棘神经元(medium spiny neuron,MSN)进行性丢失引起,NMDAR活性可被突变的htt增强[61- 63]。Shehadeh等[64]在htt及突变htt转基因小鼠上提取的纹状体MSN中给予NMDA,发现突变htt纹状体MSN凋亡程度显著增加。此外,Zhang等[65]的体内外实验表明,突变htt诱导的NMDAR可介导Ca2+内流及下游信号强度增加,指出纹状体MSN对NMDAR介导的细胞凋亡较为敏感。
肾损伤肾小球进一步过滤血液循环中的谷氨酸,并在近曲小管重吸收,当出现多种原因引起的急、慢性肾损伤时,血浆谷氨酸水平升高,NMDAR活化改变,其介导的细胞凋亡在肾损伤中起重要作用[66- 67]。
Husi等[68]对来自叶酸性肾病小鼠的肾皮质进行高分辨率蛋白质组学分析,结果表明NMDAR过度激活引起的细胞凋亡参与急性肾损伤。而后Xu等[69]研究发现,给予NMDAR抑制剂可通过降低细胞凋亡来改善大鼠缺血再灌注造成的急性肾损伤。此外Gao等[70]通过将草甘膦诱导的肾近曲小管上皮细胞进行MK- 801预处理,发现NMDAR的抑制减弱了细胞中ROS的增加,并显著降低细胞凋亡。
糖尿病-β细胞功能异常胰岛中β细胞易受胞外谷氨酸浓度影响,抑制NMDAR及其介导的细胞凋亡可延迟或阻止β细胞破坏,进而阻止糖尿病进展,甚至预防或逆转其表现[71- 72]。
Marquard等[73]研究发现,与糖尿病小鼠相比,给予NMDAR拮抗剂氢溴酸右美沙芬(Dextromethorphan,DXM)的糖尿病小鼠胰岛中,裂解型Caspase- 3蛋白表达及凋亡细胞的数量较低,表明在糖尿病中抑制NMDAR可增强β细胞存活率。Huang等[74- 75]体内外实验重复验证了此观点,并用NMDA持续刺激MIN6细胞,致胞内Ca2+和ROS浓度升高,线粒体氧化磷酸化表达降低,Bim、Bax上调,Bcl- 2下调,提出NMDAR可通过线粒体途径诱导β细胞凋亡。
视网膜损伤谷氨酸为视网膜重要兴奋神经递质,NMDAR可介导多种视网膜细胞凋亡,加重视网膜疾病进展[76- 78]。Kwong等[79]向成年新西兰白兔眼球玻璃体内注射NMDA,发现视网膜神经节细胞(retinal ganglion cell,RGC)凋亡率显著增加,且有报道指出,在NMDA诱导大鼠视网膜细胞凋亡过程中,calpain水平有所提高[80]。Gu等[81]研究发现,MK- 801可改善慢性高眼压大鼠RGC凋亡。最近一项研究在大鼠眼球玻璃体内分别注入NMDA和PBS,结果发现注入NMDA后3 h的大鼠视网膜中RGC数量显著降低,核因子κB(nuclear factor-κB,NF-κB)、p53和激活蛋白- 1(activator protein- 1,AP- 1)表达显著增加,表明NMDAR通过激活NF-κB、P53和AP- 1介导了大鼠RGC细胞凋亡,进而引起视网膜损伤[82]。
小结与展望
综上,NMDAR在细胞凋亡进程中具有重要作用,可通过线粒体及内质网、NOS、MAPK、calpain等多种途径诱导细胞凋亡。抑制NMDAR的激活,可减轻或阻止疾病对细胞、组织及器官的进一步损伤,减少死亡率与致残率。NMDAR组成及功能复杂,在不同分布类型中可介入各自级联反应,因此结合位点的识别与特异性活性药物的研究可作为该通路进一步的研究方向。选择性靶向NMDAR及其下游的多种促凋亡途径或将成为疾病发展中阻止细胞凋亡进程且耐受良好的治疗策略。此外,在NMDAR的靶点药物研究上,可以发挥中医药多成分、多靶点的作用特点,结合该通路及下游途径抑制细胞凋亡的机制进行探索。
