Non-alcoholic fatty liver disease: Epidemiology, pathophysiology and an update on the therapeutic approaches
2022-03-17RehabAbdelRahman
Rehab F. Abdel-Rahman
Department of Pharmacology, Medical Research and Clinical Studies Institute, National Research Centre, 12622- Dokki, Giza, Egypt
ABSTRACT
Non-alcoholic fatty liver disease (NAFLD) denotes a spectrum of fatty liver disease in individuals without significant alcohol consumption. NAFLD is set to be the most common etiology of serious liver diseases in numerous nations when accompanied by obesity and type 2 diabetes. It is further histologically categorized into the non-alcoholic fatty liver (NAFL; steatosis without hepatocellular injury) and non-alcoholic steatohepatitis (NASH)which is characterized by the coexistence of hepatic steatosis and inflammation and is accompanied by hepatocyte injury(ballooning), either with or without fibrosis. NAFL is considered the benign and reversible stage arising from the excessive accumulation of triglycerides in hepatocytes. However, NASH is a more progressive stage of NAFLD, due to the increased risks of evolving more serious diseases such as cirrhosis, hepatocellular carcinoma. This concept, however, has been lately challenged by a hypothesis of multiple parallel hits of NAFLD, in which steatosis and NASH are separate entities rather than two points of the NAFLD spectrum, not only from a set of histological patterns but also from a pathophysiological perspective. The current review highlights the epidemiology and pathophysiology of NAFLD, and its progression towards steatohepatitis, with special focus on the novel imminent therapeutic approaches targeting the molecular aspects and the pathogenic pathways involved in the development,and progression of NAFLD.
KEYWORDS: Non-alcoholic fatty liver disease; Steatosis;Insulin sensitizers; Glucagon-like peptide-1 analogues; Dipeptidyl peptidase-4 inhibitors; Farnesoid X receptor agonists
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) is an increasing global public health issue and is a common cause of chronic liver disease.Terminologies such as fatty-liver hepatitis, diabetes hepatitis,non-alcoholic Laennec’s disease, and alcohol-like liver disease were identified to describe NAFLD[1]. As stated by the American Association for the Study of Liver Diseases, the incidence of fatty liver in individuals without prior history of chronic high alcohol intake is referred to as NAFLD[2].
The entire disease spectrum of NAFLD could be histologically categorized into two phenotypes: non-alcoholic fatty liver (NAFL)and non-alcoholic steatohepatitis (NASH)[3]. Of note, the hallmark feature of NAFLD is steatosis wherein excessive intrahepatic triglyceride (TG) content >5% of liver weight or liver volume, or histologically identified when 5% or more of hepatocytes having apparent intracellular TG[4]. However, NASH is defined as the presence of hepatic steatosis and inflammation with evidence of hepatocyte ballooning with or without scarring or fibrosis[5]. As illustrated in Figure 1, albeit many NAFLD cases are benign in prognosis, however, it is predicted that some cases will progress from simple steatosis to NASH, fibrosis, cirrhosis, and even hepatocellular carcinoma, therefore the discrepancy between NAFL and NASH is important[6].

Figure 1. Scheme of non-alcoholic fatty liver disease (NAFLD). NAFL: non-alcoholic fatty liver; NASH: non-alcoholic steatohepatitis.
NAFLD is highly accompanied by metabolic comorbidities,like overweight/obesity, dyslipidemia, type 2 diabetes (T2DM),augmented visceral adiposity, and hypertension[7,8]. Hence, it is of significance to understand the pathophysiological mechanisms underlying the development of NAFLD.
NAFLD is supposed to be the hepatic manifestation of metabolic syndrome (MetS). It could be recognized as the presence of 3 or more of the following manifestations: (1) abdominal obesity(waist circumference >102 cm in men, >88 cm in women), (2)increased fasting glucose levels (>110 mg/dL), (3) low highdensity lipoprotein (HDL) levels (<40 mg/dL in men, <50 mg/dL in women), (4) elevated fasting plasma TG (>150 mg/dL), and (5)high blood pressure (>130/80 mm/Hg)[5].
Despite being a common health issue, NAFLD remains unrecognized and undiagnosed since most patients with NAFLD are asymptomatic. NAFLD is typically recognized when imbalanced liver biomarkers are attained on routine laboratory investigations.
It is noteworthy to mention that, an old “two-hit” theory was suggested years ago explaining that insulin resistance (IR)represented as the ‘‘first hit’’ by inducing lipid accumulation in the hepatocytes, and a second “hit” was required to develop NASH from NAFL, for instance, oxidative stress. Yet, this theory is now outdated and it is believed to be “multifactorial”, in which many factors and molecular pathways contribute to the progression of NAFLD into NASH[9].
Notably, loss of weight and lifestyle modification should be taken into account as the first-line therapy of NAFLD. Hereafter,the current treatment and management modalities are concerned with correcting the underlying metabolic disorders associated with NAFLD through insulin-sensitizing agents, lipid-lowering agents,and avoiding hepatotoxic drugs, if any[10].
In the current review, a summary of the existing therapeutic strategies up to date and the future therapeutic approaches in correlation with the pathophysiological mechanisms for NAFLD will be underlined.
2. Epidemiological features and risk profiling of NAFLD
NAFLD has progressively become one of the main diseases accountable for liver insufficiency, hepatocellular carcinoma,and liver transplantation[11]. There is an increasing incidence of NAFLD that has been correlated with the prevalence of global obesity and other metabolic complications, including diabetes,hypertension, and dyslipidemia. Thus, there is a critical need to identify the risk factors for NAFLD in order to lessen the morbidity and mortality associated with this serious illness.
2.1. Prevalence of NAFLD
NAFLD represents the main etiology of liver disease in Western nations; however, its distribution is emerging in developing countries as well. The global prevalence of NAFLD is highly reported in the Middle East (32%), South America (31%) and followed by Asia (27%), USA (24%), and Europe (23%), however,it is less common in Africa (14%)[12].
NAFLD affects about 20%-30% of the world population.The estimated prevalence of steatosis is 45% in the Hispanic population, 33% in the Caucasian population, and 24% in the African-American individuals. With regard to gender differences,the prevalence is about 42% for men and 24% for women[13].Remarkably, children are recognized to be at risk of developing NAFLD as well: the prevalence is almost 3%-10% in lean children and around 53% in obese pediatric populations[14].
2.2. Risk factors for NAFLD
The major risk factor for NAFLD and NASH is the coexistence of MetS. Obesity, IR, hypertension, hyperglycemia, and hyperlipidemia are the main features of MetS. The link between NAFLD and characteristics of MetS is bidirectional, meaning that MetS holds the risk of NAFLD, as well as NAFLD, augment various features of MetS[15].
2.2.1. Linking between obesity and NAFLD
The incidence of NAFLD among obese people is ranging between 30%-37%. A strong correlation between abdominal obesity and increased waist circumference in NAFLD is present[10]. A study indicated that 80% of NAFLD subjects are obese with body mass index (BMI) >30 kg/m2as well as, high visceral adipose tissue is found in morbidly obese persons (BMI >40 kg/m2) which leads to high NAFLD morbidity[16].
2.2.2. Role of diet composition to NAFLD
Supporting evidence that NAFLD is correlated with obesity, thus,some macro- and micronutrients implicated more to the prevalence of NAFLD. In this aspect, excessive carbohydrate intake is strictly associated with the incidence of NAFLD, such as fructose, which is a major player. It can be proposed that sugars stimulate de novo lipogenesis (DNL) and evoke an inflammatory response leading to hepatocytes apoptosis, mediated by the c-Jun-N-terminal pathway[10].
2.2.3. Diabetes as a risk factor for NAFLD
NAFLD prevalence in patients with T2DM is greater than in the general population. It is estimated that about 50%-70% of diabetic people have NAFLD. Also, NAFLD is believed to be an evolving risk factor for T2DM and a contributor to arising chronic diabetesrelated complications[17]. As well, T2DM and IR contribute to adipose tissue lipolysis and subsequent release of free fatty acids (FFAs) and further deposited in the liver evolving hepatic steatosis[18].
2.2.4. Genetic predisposition to NAFLD
Heritability and inter-ethnic variations in vulnerability propose that genetic factors can act a significant role in defining the phenotypic manifestation and overall risk for NAFLD. For example, NAFLD is present in families having genetic variants on or near NCAN, PNPLA3, and TM6SF2 that increase NAFLD heritability chance by up to 27% within families[19].
2.2.5. Gender and age-related risk for NAFLD
In general, there are sex differences in NAFLD, where the progression of NAFLD and NASH is higher in men. Indeed,women at their reproductive period are at a lower risk of NAFLD compared with men, whilst women after menopause miss this protective effect and show an equivalent prevalence of NAFLD as men[10].
2.2.6. Racial/ethnic differences in NAFLD
Another variable affecting NAFLD prevalence is race/ethnicity,with the uppermost prevalence among Hispanics, and the lowest prevalence is in African Americans. Largely, genetic and lifestyle differences among these ethnicities might be accountable for the observed differences in the prevalence of NAFLD[19].
2.2.7. Sleep deprivation as a risk factor for NAFLD
In this era, sleep disorder is a common medical problem.Epidemiological studies have proved that impaired sleep quality and sleep deprivation are correlated with the incidence of obesity,which offers a fundamental role in the pathogenesis of NAFLD.The biological evidence for this issue has been assured by the increased inflammatory cytokines levels of tumor necrosis factor-α(TNF-α) and interleukin 6 (IL-6), which in turn result in increasing fat lipolysis causing hepatic overflow of FFAs. Moreover, sleep deprivation can influence the hypothalamus-pituitary-adrenal axis, and consequently affects cortisol metabolism resulting in the accumulation of fat in the liver[10].
2.2.8. NAFLD in non-obese or lean individuals
Albeit NAFLD is highly related to MetS and obesity, however,about 5%-8% of NAFLD patients in the Western countries are lean. Remarkably, a high ratio of lean individuals with NAFLD are having PNPLA3 rs738409 GG genotype with low adiponectin serum levels, comparable to the controls. These facts denote that lean NAFLD individuals have imbalanced glucose metabolism and excess abdominal adipose tissue[12].
3. Pathogenesis and molecular basis of NAFLD
This section sheds light on the common pathophysiological and molecular mechanisms of NAFLD and the possible factors promoting its progression from simple steatosis to NASH.
The historical ‘‘two-hit’’ pathogenic theory has been switched to a multifactorial model “multiple parallel hits theory”, which recapitulates more precisely the complex and multifaceted features of the disease pathogenesis. Day and James first proposed the“two-hit” hypothesis in 1998[20]. Basically, the ‘‘first hit’’ occurs by inducing hepatic steatosis via lipid accumulation in the hepatocytes owing to IR, which increases the liver vulnerability to numerous probable “second hits” that subsequently initiate developing inflammation, fibrosis, and cellular death characteristics of NASH.The second hit can be a diversity of factors, like oxidative stress,proinflammatory cytokines, endoplasmic reticulum (ER) stress, and gut-derived bacterial endotoxin. Yet, this theory is now outdated and it is believed to be “multifactorial”, in which many factors and molecular pathways contribute to the progression of NAFLD into NASH[21].
Liver steatosis is the hallmark feature of NAFLD histological diagnosis. Different mechanisms may lead to fatty liver or steatosis(Figure 2), such as (1) high fat supply through a high-fat diet and excessive lipolysis; (2) declined FFA β-oxidation; (3) reduced export of very-low-density lipoprotein (VLDL)-TG and; (4)elevated DNL[22]. The molecular mechanisms accountable for fat accumulation in the liver are not entirely realized; nevertheless,some cytokines aroused from sites of inflammation, mostly from extrahepatic adipose tissues, could evoke these processes.Obviously, augmentation of hepatic DNL is believed to be a unique feature of the fatty liver. Notably, IR appears to be in the limelight for the metabolic dysregulations that induce hepatic steatosis[23].
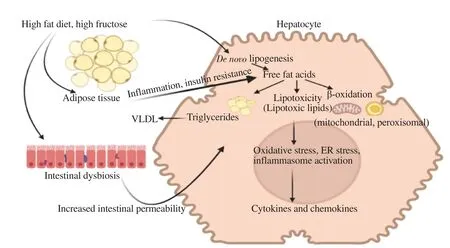
Figure 2. Different mechanisms leading to fatty liver. VLDL: very-low-density lipoprotein; ER: endoplasmic reticulum.
3.1. Adipose tissue inflammation
It is uncertain what definitely initiates adipose tissue inflammation in obesity; however, hypoxia and death of rapidly expanding adipocytes are thought to be involved. Adipocytes under inflammation secrete chemokines and cytokines, especially IL-6,TNF-α , and CC-chemokine ligand-2. Serum levels of inflammatory cytokines remarkably correlate with the existence of IR. CC-chemokine ligand-2 recruits macrophages to the adipose tissue,bringing yet further local production of cytokines, and continuation of the inflammatory cycle[24].
IL-6 and TNF-α initiate a state of adipocytes IR, which induces TG lipolysis and release of FA into the circulation. Together, extrahepatic adipocytes are challenged in their innate capability to adiponectin secretion, an anti-inflammatory adipokine that augments the normal degradation of lipid to adipocytes for storage. The circulating adiponectin regulates fatty β-oxidation in the liver via acetyl-CoA carboxylase and 5’ adenosine monophosphate-activated protein kinase (AMPK) signaling. Together, these irregularities stimulate loss of fat from adipocytes and promote ectopic fat accumulation[25].
3.2. DNL
Intrahepatic accumulation of fat is a consequence of the disbalance between lipid acquisition and disposal that are organized via four main pathways: circulating lipids uptake, fatty acid oxidation (FAO), DNL, and lipids export as VLDL. Accordingly,fat accumulation because of lipid acquisition pathways exceeds the disposal pathways[26].
Carbohydrates are substrates for the DNL, accordingly, the amount of carbohydrates in diet could be directly be correlated with DNL in the liver. Simple sugars are converted to FAs easier than complex carbohydrates, and fructose is an efficient inducer of DNL than glucose. Notably, dietary fat, especially saturated fat,evokes DNL by upregulating sterol responsive element binding protein (SREBP)-1, a principal regulator of the hepatic lipogenic genes[27]. DNL allows the liver to produce new fatty acids from acetyl-CoA. The newly synthesized FA may then experience a range of desaturation, elongation, and esterification steps before final storage as TG or exported as VLDL particles[26].
3.3. IR
IR is a characteristic feature of NAFLD, however, not all people with NAFLD have increased IR. IR is triggered by several factors, involving soluble mediators originated from immune cells and/or adipose tissue, for instance, IL-6 and TNF-α. Serine phosphorylation of insulin receptor substrate-1 (IRS-1), is one of the key aspects that disrupt signaling of insulin, via either the inflammatory signal transducers for instance c-jun N-terminal protein kinase 1 (JNK1) or nuclear factor-κB kinase-β (IKK-β)inhibitor. Additionally, individuals with NAFLD and IR show decreased insulin sensitivity, not only at the muscle level but also in the adipose tissue and liver, which result in more complex metabolic disturbance of lipid and glucose[28,29].
3.4. Lipotoxicity
Actually, in NAFLD, some lipids might be harmful to hepatocytes. It is known that dairy products and animal fat are rich in long-chain saturated fatty acids (SFAs) like stearate and palmitate. Normally, SFAs are transported to the mitochondria for esterification or β-oxidation for either being excreted as VLDL or to be stored as fat droplets. Diverse mechanisms are engaged in liver injury, which is emphasized by SFAs and by free cholesterol from de novo synthesis. Besides, accumulation of free cholesterol in hepatocytes results in hepatic injury via intracellular activation of signaling pathways in hepatic stellate cells, Kupffer cells, and consequently, promotes inflammation and fibrogenesis[30].
3.5. Oxidative stress and mitochondrial dysfunction
The imbalance between the limited antioxidant defense system and exaggerated reactive species production as reactive nitrogen species (RNS) or reactive oxygen species (ROS) gives rise to oxidative stress. In fact, mitochondria are a major source of ROS generation in the cells. FFAs are metabolized via the tricarboxylic acid cycle and the mitochondrial β-oxidation pathways, which in turn generate citrate and inhibit glycolysis. Consequently, reduced oxidation of glucose and uptake in skeletal muscle through glucose transporter type 4. In obese individuals, elevated FFA uptake in the liver augments FA hepatic oxidation as compensation for the excessive hepatic fat storage. Mitochondrial FFA oxidation is kept till mitochondrial respiration turns severely compromised. Besides,accelerated β-oxidation results in high production of ROS, and may lead to mitochondrial dysfunction[28].
3.6. ER stress
ER has a vital role in protein synthesis, folding, and trafficking.Under stress, like hypoxia, inflammation, and energy excess, as in NAFLD, ER effectiveness is decreased. ER dysfunction leads to the accumulation of unfolded proteins thereafter, triggering unfolded protein response (UPR), as an adaptive response to resolve ER stress. UPR is characterized by amplified proteins degradation and translational stoppage of protein synthesis to restore the normal functions of ER. Additionally, UPR mediates both, the immune and metabolic responses that exacerbate IR. One of the insults of ER stress is obesity leading to insulin signaling suppression via serine phosphorylation of IRS-1 and stimulation of the JNK pathway[31].
3.7. Intestinal microbiome and mechanisms of NAFLD
Altered gut flora was first traced over 80 years ago, in individuals with chronic liver disease. It was found that, in about 20%-75% of individuals with chronic liver disease, imbalance of the gut microbiota, particularly the small intestinal bacterial overgrowth,highly exists[29].
Dysbiosis is a disturbance in the composition of the microbiota,which can be a loss of beneficial microbiota, decreased microbiota variety, or increased pathogenic microbiota. Under dysbiotic conditions, disruption in the intestinal barrier arises, as the gut microbiota is unable to sustain its role in local intestinal homeostasis[32]. Based on evidence from related researches, the gut microbiota could contribute to NAFLD pathogenesis via multiple mechanisms, as (1) elevated production and absorption of short-chain FAs in the gut; (2) alteration in bile acid pools by the microbiota; (3) alteration in the metabolism of dietary choline by the microbiota; (4) elevated delivery of microbiota-derived ethanol to the liver; and (5) alterations in gut permeability and leakage of endotoxin[29].
To conclude, similar to other metabolic disorders, the molecular pathways regulating the incidence and aggression of NAFLD are complex. Despite an accurate understanding of lipid metabolism nowadays, a thorough knowledge of factors governing the transition from fatty liver to NASH is needed.
4. Preclinical models of NAFLD
In this section, the most frequently used animal models for NAFLD referring to their mechanism of NAFLD pathogenesis in each model are highlighted.
4.1. Dietary animal models of NAFLD
4.1.1. Nutrient-deficient NAFLD models
Numerous preclinical animal models exist, relying on deficient diets for specific essential nutrients to induce a liver phenotype simulating human NAFLD.
4.1.1.1. Methionine and choline-deficient (MCD) diets
The MCD diet contains elevated fat (10%) and sucrose (40%)but is lacking both methionine and choline. Obviously, the MCD diet induces steatosis and inflammation rapidly in the hepatic tissues, subsequently tissue injury and fibrosis within a few weeks of feeding. It is documented that methionine and choline are basically methyl group donors, and adequate methylation capacity is required for maintaining hepatic lipid metabolism[33].
很快,扎扎的细响就被头顶花朵中间传来的声音盖住了。好像有风刮起来,卷过平原上的麦田,山岭上的松林,又吹开东海上的巨浪,由青萍之末到飞沙走石,到激扬澎湃,直至浩荡往复,好像能够将星斗一颗颗吹落到大地上。但这并非是真正的风,袁安手中的火把熊熊燃烧,纹丝不动,他们的头发也没有被吹起。
4.1.1.2. Choline-deficient diet
In fact, a choline-deficient diet is mostly folate-deficient too. Lately,studies proposed that a low intake of choline can evoke NAFLD progression in humans. Comparable to choline and methionine, folate is a third essential methyl group donor in hepatocyte metabolism and some clinical studies depicted a relationship between folate deficiency and the severity of NAFLD[34].
4.1.1.3. Choline-deficient L-amino-acid (CDAA) diet
CDAA diet is parallel to the MCD diet, both are deficient in choline, but the L-amino acids mixture substitutes equivalent proteins. CDAA diet-fed mice have increased inflammation,oxidative stress, lipid synthesis, and resulting in liver fibrosis[35].
4.1.2. Obesogenic high-fat diet models of NAFLD
4.1.2.1. High cholesterol diet
High level of blood cholesterol is a well-established risk factor for developing NAFLD. In line with this perception, the liver of the high-fat diet (HFD)-based mouse model can be intensified by increasing cholesterol in the diet[35].
4.1.2.2. High-fructose diet
Increased dietary intake of fructose is an important risk factor for the development of NAFLD. High fructose is metabolized in the liver by fructokinase C leading to adenosine triphosphate depletion and generation of uric acid. Unfavorably uric acid increases hepatic TG accumulation and inflammasome activation. Besides, studies disclosed the crucial involvement of fructose in intestinal dysbiosis and the activation of hepatic DNL[35].
4.2. Genetic animal models of NAFLD
4.2.1. Leptin-deficient mice (ob/ob)
Ob/ob mice are lacking functional leptin, a satiety hormone, and so they are hyperphagic, develop hyperlipidemia, obesity, and IR on a standard chow diet. Ob/ob mice do not advance to NASH till challenged by further metabolic stimuli, for instance, subjected to minor doses of lipopolysaccharide or fed with particular chows,such as MCD diet or HFD[36].
4.2.2. Leptin receptor-deficient mice (db/db)
There are physiological similarities between db/db mice and ob/ob mice in having a spontaneous mutation in the db gene encoding leptin receptor. Similar to ob/ob animals, db/db mice spontaneously develop steatosis under regular nutritional conditions. They necessitate further stimuli to reveal disease progression into NASH[36].
4.2.3. Low-density lipoproteins (LDL) receptor-deficient mice (Ldlr-/-)
Receptors for LDL (LDLR) are cell surface receptors expressed on many cell types mediating endocytosis of cholesterol-rich LDL particles. Derangement of that pathway as in LDLR-deficient mice(Ldlr-/-) results in impaired circulatory clearance of cholesterol[34].
4.2.4. Foz/foz mice
Foz/foz mice are obese and possess a mutated Alms1 gene, they spontaneously reveal liver steatosis, obesity, and IR with elevated cholesterol levels. Likewise, feeding HFD can promote the transition into steatohepatitis[37].
4.2.5. Apolipoprotein E (ApoE)-deficient mice (Apoe-/-)
ApoE is a vital element of transporting lipoprotein particles,abundant amounts of lipids, and cholesterol. Lacking ApoE in mice gives rise to impaired lipoproteins clearance from the circulation.Nourishing Apoe-/- mice with a high-fat/high-cholesterol diet for 7 weeks resulted in steatosis[34].
4.3. In vitro models of NAFLD
Several in vitro models of NAFLD have been developed, mainly consisting of primary hepatocyte cultures or hepatocyte cell lines treated with monounsaturated and/or saturated FAs (MUFAs and SFAs). These models usually employ palmitate or oleate, or both,as common long-chain FAs in the Western diet and as the most abundant FAs in the liver of normal and NAFLD individuals[7].
4.3.1. Rat hepatocyte primary cultures
Rat hepatocyte primary cultures have been widely used in basic research on liver function. Hepatocellular steatosis in primary cultures is achieved within only 1 day of treatment with FA mixture[38].
4.3.2. Rat hepatoma cell line
A wide-ranging array of liver-specific mRNAs are expressed by FAO rat hepatoma cells. These cells sustain the capability to assemble and release VLDL as well as to respond to stimuli that stimulate peroxisome proliferator-activated receptors (PPAR).Interestingly, the basal lipid content of FAO cells is less than those in the primary hepatocytes, so, when subjected to FA, more rapid and pronounced TG accumulation aroused compared to the primary cultured hepatocytes[7].
4.4. Less commonly used models
4.4.1. Neonatal streptozotocin (STZ)/diabetes model
Intraperitoneal or subcutaneous injection of a low dose of STZ shortly after birth induces type 1 diabetes due to pancreatic islets damage. Feeding HFD diet to STZ mice for 6 to 8 weeks leads to hepatic steatosis, inflammation, ballooning, and progressive pericellular fibrosis[33].
4.4.2. Monosodium glutamate
This model is done in 3-4-months-old mice injected with a single dose of monosodium glutamate (4 mg/g b.wt.) within 5 d after birth. This results in obesity, oxidative stress, IR, T2DM,and hyperlipidemia. In the liver, marked microvesicular steatosis,centrilobular hepatocellular ballooning with Mallory-Denk body,megamitochondria, scattered neutrophil granulocyte foci, and mild perivenular fibrosis could be detected[33].
5. Current treatment strategies for NAFLD
Effective treatment of NAFLD patients is challenging due to the complex etiology, problematic diagnosis, the divergent spectrum of its stages, and the possibility for coexisting diseases as well.A number of the reported treatment strategies have designated a change in lifestyle, insulin sensitizers, lipid-lowering agents,antioxidants, and cytoprotective agents as a therapeutic measure against multifaceted pathways of NAFLD.
5.1. Lifestyle intervention
For NAFLD is correlated with an unhealthy diet, increased body weight, and physical inactivity, therefore, modification of lifestyle heading for weight loss and increased physical activity is the firstline treatment[39].
5.2. Bariatric surgery
For individuals who are not responding to lifestyle modifications and pharmacotherapies, bariatric surgery is a choice for losing weight. Thus, it is an effective solution for obese and diabetic individuals with NAFLD to induce weight loss, resolve NASH inflammation, and improve longevity[40].
5.3. Pharmacological treatment
Most guidelines acknowledged that any drugs prescribed for NAFLD should be considered as an off-label therapy and that decision should be discussed with the patient, in order to carefully balance between the benefits and the safety[41]. Table 1 summarizes some of the most commonly used pharmacologic treatments of hepatic steatosis.

Table 1. Pharmacologic therapies with benefits in NAFLD/NASH treatment.
5.3.1. Antioxidants and cytoprotective agents
Vitamin E is a lipid-soluble vitamin with antioxidant properties.It has a significant role in lipid peroxidation and membrane stabilization as chain-breaking in the free radical reactions. It stabilizes biological membranes by protecting unsaturated FAs from lipid peroxidation. Another role beyond its antioxidant effect includes attenuating cytokine stimulation of stellate cells by reducing TGF-β levels[42].
Ursodeoxycholic acid (UDCA) is one of the cytoprotective agents. It is efficacious in treating cholesterol gallstones and primary biliary cirrhosis. Its pharmacotherapeutic effects include anti-apoptotic, enhancing hepatic insulin sensitivity, lowering serum TNF-α levels, and lowering ER stress, which proposes that UDCA could be beneficial in NASH treatment. Paradoxically,UDCA reduces liver enzymes but does not show improvement of the histological features in NASH[43,44].
S-adenosyl-L-methionine (SAMe) is a conditionally essential amino acid formed from methionine and adenosine triphosphate. It is responsible for cell membrane stabilization, VLDL production,and glutathione (GSH) production[45]. As well, experimental evidence shows SAMe capability to attenuate proinflammatory TNF-α-mediated hepatic injury possibly through the downregulation of transcription factor and nuclear factor kappa-B(NF-κB)[46].
Betaine (trimethylglycine) is a naturally occurring choline metabolite. Its theoretical benefits include antioxidant effects (by increasing SAMe) as well as lipotropic effects and membrane stabilization. Moreover, betaine reverses IR by inducing IRS-1 phosphorylation along with improving the downstream pathways associated with gluconeogenesis and glycogen synthesis[47].
N-acetylcysteine (NAC) is a GSH precursor that improves hepatic GSH levels and protects against oxidative stress. Dietary NAC has been studied in an animal model with hepatic steatosis,and it resulted in abolished NASH-induced lipid peroxidation,elevated hepatic GSH, inhibited TNF-α production, diminished inflammation, a consequent reduction in hepatocyte injury, and fibrosis[48].
5.3.2. Insulin sensitizers
5.3.2.1. Thiazolidinediones (TZD)
TZDs are insulin-sensitizing compounds having the ability to ameliorate liver steatosis as well as IR, histologic and biochemical parameters[41]. TZDs are PPAR-γ agonists. They enhance insulin sensitivity via regulation of the transcription of several genes encoding for lipid and glucose metabolism. To protect the liver from the deleterious metabolic effects of FFA, TZDs decrease lipolysis and promote FA uptake for storage in adipose tissue and thus increase adipose tissue mass. Likewise, TZDs administration affects the production of adipokines that stimulates FA oxidation and inhibits lipid accumulation. Thus, adiponectin could possibly be considered one of the key mediators in TZDs-decreased accumulation of fat in hepatocytes. Besides, TZDs decrease TNF-α,which is positively related to the degree of steatosis and fibrosis[49].Rosiglitazone, a PPAR-γ agonist, affects adipogenesis and hepatic fat homeostasis via favoring of fat deposition in the adipose tissue rather than in the liver. So, it decreases the hepatic TG content in NAFLD. Both rosiglitazone and pioglitazone have been broadly studied in patients with NAFLD; yet, in 2010 rosiglitazone was withdrawn from the European market due to a high risk of myocardial infarction[50,51].
Pioglitazone, a TZD, is considered safe for cardiovascular(CV) outcomes. It is a PPAR-γ agonist for treating T2DM after rosiglitazone was withdrawal from many countries. It improves insulin sensitivity and increases glucose utilization in peripheral tissue in T2DM patients. Besides, treatment with pioglitazone increases adiponectin serum levels, decreases insulin levels, thus alleviates hepatic steatosis, reduces hepatic lipotoxicity, and improves hepatic and peripheral IR. Desperately, the principal side effects of glitazones are increased body weight gain and incidence of bone fractures in women. Likewise, the use of pioglitazone has been amended in some European countries due to the slight possibility of increased risk for developing bladder cancer after long-term use. Thus, pioglitazone use for the treatment of NAFLD is permitted by both the National Institute for Health and Care Excellence and American Association for the Study of Liver Diseases guidelines with limitations[41,52].
Saroglitazar is another insulin sensitizer, with a dual PPAR α/γ agonist used for treating diabetic dyslipidemia. Furthermore, it revealed promising effects in NASH experimental models and it seems to be effective in humans too, by improving serum alanine aminotransferase (ALT) levels and reducing liver size assessed in NASH patients after a 24-week course. Unlike pioglitazone,saroglitazar does not appear to be associated with peripheral edema or weight gain[51]. Furthermore, based on both in vitro and in vivo experimental results, saroglitazar reduces the expression of proinflammatory cytokines such as TNF-α, IL1-β, and IL-6. It also reduces oxidative stress, UPR, and fibrogenic signaling[53].
5.3.2.2. Metformin (MET)
MET, a biguanide, alleviates IR and hyperinsulinemia. According to the American and European guidelines, it is the first-choice pharmacotherapeutic agent for T2DM. It is an insulin sensitizer that inhibits lipolysis, and the subsequent FFA release from the adipose tissue reduces hepatic gluconeogenesis and enhances the oxidation of FA. MET also augments glucose uptake and storage from the muscle, and decreases glucose absorption from the intestine, and thus improves insulin sensitivity[51]. Moreover,several studies revealed that MET normalizes transaminases and suppresses hepatic steatosis; though, histologic data remain limited[28].
5.3.3. Lipid-lowering agents
5.3.3.1. Statins
Statins are powerful lipid-lowering agents that diminish the risk of CV events in patients with coronary artery disease and diabetics.The therapeutic effects of statins are owed to their cholesterollowering action, via diminishing cholesterol biosynthesis through inhibiting 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase.Besides, statins have also been reported to have anti-inflammatory,antioxidative, antifibrotic, antithrombotic, and immunomodulatory actions, thus proposing statins as a therapeutic option for patients with either NAFLD or NASH[54]. Added to that, statins improve the peroxisomal and mitochondrial FA oxidation via increasing PPAR-α gene expression. Likewise, statins stimulate hepatic LDL receptor-related protein 1 (LRP-1) that reveal an important role in the clearance of circulating TG-rich lipoprotein remnants,chylomicrons, and VLDL in the liver. Albeit physicians usually avoid prescribing statins for patients with NAFLD, their use has been found to increase liver enzymes and decrease CV morbidity and mortality. So, the risk of not taking statins will likely outweigh the risk of taking it in those patients[41]. Two subclasses of statins with different functions are identified according to their bioavailability, which are hydrophobic and hydrophilic statins.Atorvastatin, simvastatin, lovastatin, and fluvastatin are lipophilic statins and are metabolized by the cytochrome P450system, and thus could reach a higher concentration in the liver compared to hydrophilic statins. Whereas pitavastatin and pravastatin are hydrophilic in nature and thus are minimally metabolized in the liver, however, rosuvastatin has an intermediate behavior.All statins seem to be efficient in lowering cholesterol levels in NAFLD patients, though atorvastatin is the only statin that revealed decreased CV events in those patients[55].
5.3.3.2. Fibrates
Fibrates are a type of amphipathic carboxylic acids, owning both hydrophilic and lipophilic properties. They are a much-used class of lipid-modifying agents resulting in an extensive decrease in plasma TG and are usually associated with a moderate decrease in LDL and an increase in HDL. Studies indicated that their effects are mediated, at least through alterations in the transcription of genes encoding for proteins controlling lipoprotein metabolism.Fibrates activate specific transcription factors, PPARs. The PPAR-γ form mediates fibrate action on HDL cholesterol levels via transcriptional induction of synthesis of the major HDL apolipoproteins constituents, apoA-Ⅰ and apoA-Ⅱ[56].
Fenofibrate results in elevated PPAR-γ levels, enhanced expression of genes promoting hepatic FA β-oxidation, and reduced hepatic steatosis. PPAR-γ activation is involved in lipoprotein metabolism by increasing lipolysis, therefore decreasing TG-rich particles. Furthermore, fenofibrate reduces hepatic IR. It also downregulates the expression of inflammatory mediators involved in NAFLD/NASH pathogenesis[57].
Gemfibrozil, another fibrate, has antihyperlipidemic effects, as it improves dyslipidemia associated with NAFLD. It decreases the production of VLDL and TG, reduces ALT without affecting liver histology, and enhances the hepatic microcirculatory system function by facilitating FA oxidation[58].
5.3.3.3. Ezetimibe
Ezetimibe belongs to a class of hypolipidemic agents that inhibits the reabsorption of lipids from the intestinal lumen into enterocytes. Moreover, it reduces hepatic lipid content, serum TNF-α, and ALT levels in a mouse model of NAFLD. Human studies for ezetimibe are anticipated[59].
5.3.4. Renin-angiotensin system blockers
The principal roles of the renin-angiotensin system in developing NAFLD have been widely examined, via targeting the mechanisms of IR and hepatic injury, for instance, telmisartan and losartan, the most common angiotensin Ⅱ receptor type 1 blockers[60].
Losartan showed significant improvement in ALT levels and serum markers of fibrosis in hypertensive patients with NASH. As well, losartan has been reported to reduce the number of activated hepatic stellate cells, which play a fundamental role in developing hepatic fibrosis, suggesting that losartan might be efficient for NASH therapy[60].
Telmisartan is another angiotensin receptor blocker. Telmisartan is expected to have more potent effects in NAFLD than those of losartan via PPAR-γ activation, which promotes hepatic FA oxidation,decreases hepatic lipogenesis, and increases peripheral and hepatic insulin sensitivity. In fact, telmisartan has been reported to improve IR and liver injury, based on serum levels of ALT and homeostasis model assessment-IR in humans. Mild headache, dizziness, and abdominal pain are the most common adverse effects[61].
5.3.5. Drugs modulating hepatocyte injury via blockade of TNF-αα
Pentoxifylline is a methylxanthine compound, a non-selective phosphodiesterase inhibitor that produces vasodilatory effects and reduces the production of oxygen-free radicals. It inhibits TNF-α and is assigned for the treatment of liver steatosis as it plays a central role in turning to steatohepatitis. However, further researches are necessary to verify the effectiveness of pentoxifylline regarding the histologic improvement in NAFLD[28].
6. New therapeutic candidates for NAFLDα
A summary of some ongoing clinical trials of pharmacotherapies for the treatment of NAFLD/NASH is listed in Table 2. Other therapeutic agents are described in the following section.
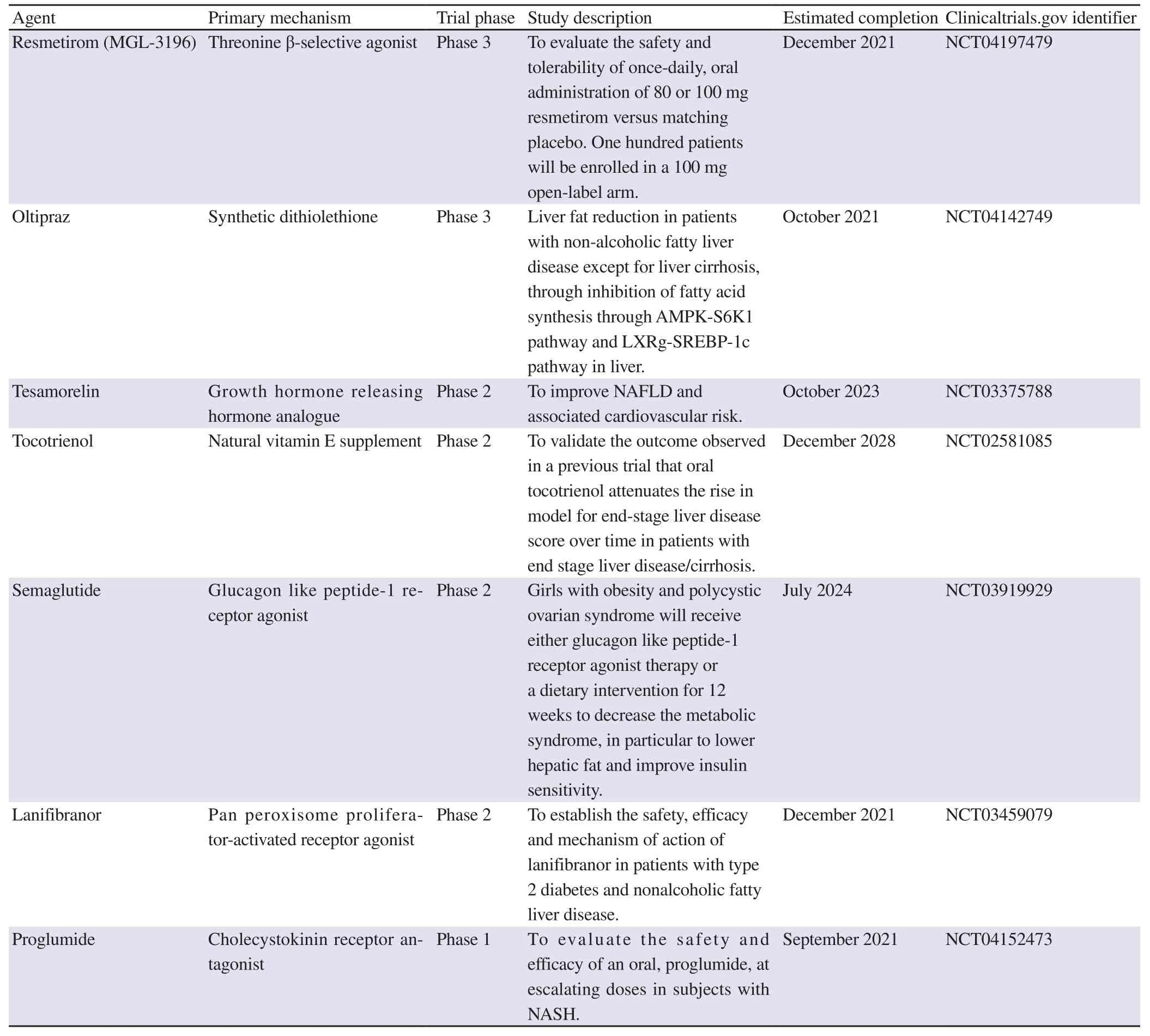
Table 2. Some pharmacotherapies undergoing clinical trials for the treatment of NAFLD/NASH.
6.1. Cilostazol
Recently, cilostazol, a selective type Ⅲ PDE inhibitor, has been shown to inhibit the expression of SREBP-1c, which is a principal regulator of the expression lipogenic gene in the liver. Experimental data revealed that cilostazol stimulates the hepatocytes’ LDL receptor-related protein promoter activity, resulting in increased expression of LDL receptor-related protein in the liver. Moreover,cilostazol revealed potential in alleviating steatosis, however, more data on its role in NAFLD therapy are necessary[28,62].
6.2. Polyunsaturated fatty acids (PUFA) and monounsaturated fatty acids (MUFAs)
PUFAs are originating mainly in soybean, cottonseed, corn,sesame, sunflower, and safflower oils. Of note, omega-3 PUFAs(n-3 PUFAs), are suggested to enhance IR via regulating mitochondrial function or mediating anti-inflammatory effect. They also have a beneficial impact on hyperlipidemia, hypertension,endothelial dysfunction, and CV disease. On the other hand,MUFAs are comprised of olive oil. Notably, phenolic contents in MUFAs have revealed anti-inflammatory and antioxidant properties that could improve dyslipidemia and endothelial dysfunction.Further studies are awaited to explore the usefulness of MUFA and PUFA in NAFLD patients[52].
6.3. MK615
MK615 is extracted from Japanese apricots “Prunus mume” and contains several cyclic triterpenes, as oleanolic and ursolic acids.It can inhibit several inflammatory cytokines production, such as IL-6 and TNF-α by alleviating NF-κB. It is a hepatoprotective agent, as it decreases hepatic enzymes. More studies are essential to elucidate its effect in patients with NAFLD[28].
6.4. Orlistat (Xenical)
Orlistat is a synthetic hydrogenated derivative of lipostatin, a naturally occurring lipase inhibitor. It is one over-the-counter diet pill. It causes direct blockage of intestinal absorption of dietary fat, and thus enhances weight loss. Yet it doesn’t always reverse fibrosis in NAFLD/NASH patients, but it appears to correct other biochemical, and metabolic anthropometric indicators[63].
6.5. Glucagon-like peptide-1 (GLP-1) analogues
GLP-1 analogues are incretin-mimetics (metabolic gut-derived hormones stimulate decreased blood glucose), exerting pleiotropic actions. GLP-1 receptor analogues have an indirect role in NAFLD pathophysiology by increasing insulin production. They can improve hyperglycemia and decrease weight gain via acting on the central nervous system and suppressing the appetite, delaying gastric emptying time and consequently weight gain reduction.Besides, direct enhancement in insulin sensitivity by inducing cAMP production and activation of protein kinase A improves lipid metabolism and decreases oxidative stress. Two agents of these GLP-I agonists are already implemented in routine clinical practice, which are exenatide and liraglutide, along with many other agents in earlier phases of clinical development, for example,albiglutide and taspoglutide[64,65].
Exenatide is the synthesized form of exendin-4, a peptide secreted in the saliva of the Gila monster, a venomous lizard. It has glucoregulatory properties as increased glucose-dependent insulin secretion, glucose-dependent suppression of glucagon secretion, delayed gastric emptying, decrease in the absorption of carbohydrates, and increased feelings of satiety. It also maintains β cell mass via enhancing the differentiation of β cells and inhibiting apoptosis. Exenatide is the first GLP-1 receptor agonist submitted in clinical practice. In diseased individuals with T2DM, exenatide treatment improved liver enzymes and attenuated steatosis, but this study is lacking histologic validation[41].
Liraglutide is a long-acting GLP-1 analogue. It had been experimentally and clinically ameliorated NAFLD. It enhances peripheral, adipose, and hepatic IR. Besides, it also decreases DNL in patients with NAFLD/NASH[44]. Liraglutide possibly might diminish the intrahepatic fat content and HbA1c, with recovering the liver functions in patients with T2DM and NAFLD. Interestingly, it was also stated that liraglutide revealed in vitro a protective mechanism against NAFLD, that might be mediated partially by the AMPK/mTOR pathway. Besides, it decreased fatty degeneration initiated by FFAs in hepatocytes via the pathway of AMPK/SREBP-1c[66].
6.6. Dipeptidyl peptidase-4 (DPP-4) inhibitors
Incretins such as GLP-1 and glucose-dependent insulinotropic polypeptide are degraded by DPP-4, and thus their half-life is prolonged using the DPP-4 inhibitor, as depicted in Figure 3.DPP-4 inhibitors are clinically used concomitantly with other oral antidiabetic drugs, such as TZDs and MET in subjects with imbalanced glycemic control, or who demand limiting weight gain.Some inhibitors of DPP-4 enzyme, that are regulating native GLP-1 bioactivity, have been evolved. Some of these agents are available for clinical use such as sitagliptin, saxagliptin, vildagliptin,linagliptin, and alogliptin[64,65].
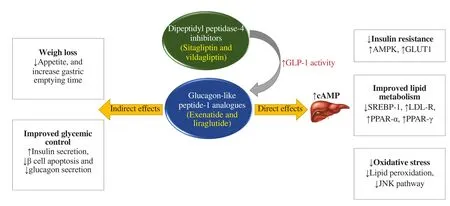
Figure 3. Adapted mechanism of action of glucagon-like peptide-1 (GLP-1) analogues and dipeptidyl peptidase-4 (DPP-4) inhibitors. AMPK: 5’ adenosine monophosphate-activated protein kinase; GLUT1: glucose transporter type 1; cAMP: cyclic adenosine monophosphate; SREBP-1: sterol responsive element binding protein-1; LDL-R: low-density lipoprotein receptor; PPAR: peroxisome proliferator-activated receptor; JNK: c-jun N-terminal protein kinase 1.
Sitagliptin, an oral antidiabetic agent, acts by increasing insulin secretion and suppressing glucagon release from the pancreas. It could be considered as a novel therapeutic agent for managing NAFLD via inhibiting DPP-4, and thus reducing lipogenesis and abolishing hepatic steatosis[44].
Vildagliptin, another DPP-4 inhibitor, acts by enhancing the incretin effect in T2DM. It could enhance weight loss, decrease the diversity of CV risk factors, protect vascular endothelial cells, and possibly has an anti-inflammatory effect. Therefore, vildagliptin is likely considered a potent agent in steatosis treatment, along with its wide use as a blood-glucose-lowering drug[67].
6.7. Glucose cotransporter 2 (SGLT2) inhibitors
It is assumed that sodium-SGLT2 inhibitors or gliflozins, such as dapagliflozin, empagliflozin, canagliflozin, and ipragliflozin,might have a role in the prevention and management of NAFLD via glucose-independent mechanisms, such as inhibiting inflammatory markers, alleviating oxidative stress, reducing lipogenesis, and enhancing FFAs oxidation (Figure 4). SGL2 inhibitors could suppress glucose reabsorption in the kidney, hence developing glucosuria. The mechanism of action of SGLT2 is independent of insulin secretion, so it could be useful in patients with limited pancreatic β cell activity. SGLT2 inhibitors also act as α-cells secretagogues through the direct neuronal stimulation of glucagon release. The mild hyperglucagonemic condition enhances hepatic gluconeogenesis, lipolysis, and ketogenesis, resulting in a reduction in the number of FAs. Besides, inhibitors of SGLT2 suppress IR resulting in downregulation of SREBP-1c and further blockage of DNL[68,69].
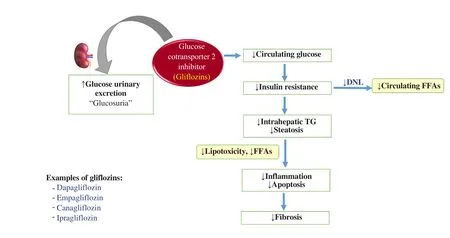
Figure 4. Adapted mechanism of action of gliflozins. DNL: de novo lipogenesis; FFAs: free fatty acids.
6.8. Farnesoid X receptor (FXR) agonists
FXRs are nuclear hormone receptors that are highly expressed in different body tissues. Moreover, FXRs regulate insulin as well as carbohydrate and lipid metabolism[44]. Preclinical studies revealed that activation of FXR could protect against fatty liver injury in NAFLD/NASH animal models. It also improved insulin sensitivity, hyperlipidemia, and glucose intolerance. Added to that, FXR agonists antagonize NF-κB signaling, resulting in anti-inflammatory activities. FXRs also downregulate SREBP1 and thereby suppress DNL. One of FXRs is obeticholic acid,which is a 6 α-ethyl derivative of the natural human bile acid chenodeoxycholic acid and is ~100-fold more potent than chenodeoxycholic acid, has been approved for the treatment of primary biliary cholangitis[70].
In June 2020, the Food and Drug Administration has rejected obeticholic acid, for the treatment of NASH fibrosis, as the predicted benefit based on a surrogate histopathologic endpoint remains uncertain and does not sufficiently outweigh the potential risks[71].
6.9. Probiotic, prebiotic, and synbiotic supplementation in NAFLD
The linkage between gut and liver is named the “gut-liver axis”,which has drawn interest in the pathogenesis of NAFLD. The gut-liver axis is associated with overgrowth of small intestinal bacteria and increased intestinal permeability. Likewise, targeting the gut microbiome is one of the promising approaches aimed at preventing hyperlipidemia and NAFLD treatment[44]. Probiotics,which are living, non-pathogenic microorganisms, improve the composition of intestinal microbiota by reducing liver inflammation and improving aminotransferases. It could improve the inflammatory factors and the metabolic parameters of NAFLD,such as total cholesterol, visceral fat, and IR. Commercialized probiotics comprised of lactic acid and spore-forming bacteria,such as Bifidobacteria and Lactobacilli[72]. On the other hand,prebiotic fibers are non-digestible carbohydrates that can be fermented by bacteria and then change the composition and activity of gut microbiota promoting health benefits. Its supplementation has been shown to reduce plasma lipids and hepatic TGs in both animals and humans, such as oligofructose[73]. Interestingly,synbiotics refer to a combination of probiotics and prebiotics that can reverse dysbiosis, besides, there is strong evidence that synbiotics are useful for attenuating inflammation among NAFLD patients. It could improve IR through mechanisms such as modifying the gut microbiome, increases in fecal pH, reductions in endotoxin concentrations, and reducing intestinal toxins production and absorption[74].
7. Natural products as therapeutic candidates for NAFLD
Because of the potential adverse effects of conventional pharmacological therapies, several studies are focusing on finding complementary therapies from natural sources acting as safe products, such as herbal medicine and functional foods.Furthermore, natural compounds supplementation is used to complement a healthy diet in order to improve NAFLD, due to their anti-steatotic and antioxidant effects[75].
Among these, Crocus sativus L. (saffron) is a perennial herb member of the Iridaceae family. Saffron, the dried stigma of the flower, is used as a flavoring spice. It is increasingly gaining attention due to its content of many bioactive molecules with health-promoting actions, such as crocin, crocetin, picrocrocin,and safranal[76]. Several studies revealed that the hepatoprotective effects, anti-oxidants, and anti-inflammatory effects of saffron could be valuable in various forms of liver injury such as viral hepatitis, drug-induced liver damage, and alcoholic and nonalcoholic steatohepatitis[77]. Saffron increases the blood flow in certain tissues and has oxidant and anti-inflammatory activities, as well as shows beneficial effects over lipid metabolism and IR[78].Additionally, there is evidence that in experimental hyperlipidemia models crocin has a hypolipidemic effect through the significant decrease in serum TGs and LDL levels, besides the inhibition of pancreatic lipase activity[75].
Moreover, Curcuma longa L. is used in the Far East folk medicine for the treatment of biliary disorders[79]. Curcumin, which is the active principle of turmeric (extract of Curcuma longa L.),is known for reducing inflammation and alleviating oxidative stress. Likewise, curcumin protects the liver from obesity-induced steatosis and could improve mitochondrial function[80].
Interestingly, Capsicum annuum L. (Solanaceae family), or hot pepper, contains capsaicin, which is responsible for pungent properties. Capsaicin administration decreases fat-induced inflammation in adipose tissue by suppressing the production of IL-6, TNF-α, monocyte chemoattractant protein-1, and cyclooxygenase-2 and it increases adiponectin expression[81].
There is accumulating evidence showing that Glycyrrhiza glabra L. (licorice) that belongs to the Fabaceae family, has anti-inflammatory, anticancer, antioxidant, and antimicrobial effects[82]. The primary constituent of licorice is glycyrrhizin which is a triterpene glycoside showing protective activity on hepatic disorders. Furthermore, the anti-steatotic effect of licorice flavonoid oil has been evidenced by a reduction of abdominal adipose tissue and plasma cholesterol and TGs levels, with a reduction of acetyl CoA carboxylase and an increase in the catabolic acyl-CoA dehydrogenase[83].
Of note, Allium sativum L. (garlic) is widely used in cooking and it also has therapeutic properties. The hepatoprotective activities are evidenced by the decrease in ALT, aspartate aminotransferase,and alkaline phosphatase levels, increased hepatic hexokinase and G6Pase activity, with the consequent reduced hepatic glycogen content, and decreased lipid peroxide levels, resulting in a significant decrease of hepatic steatosis in diabetic animal models[75].
Notably, garlic stem contents of chlorophyll, carotenoids, and vitamin C at higher levels are accountable for its antioxidant activities. Allium sativum stem extract could normalize adiponectin and leptin levels, and inhibit fat accumulation in obese mice by modulating the activities of hepatic lipid regulating enzymes and suppressing GSH depletion and lipid peroxidation in hepatic tissue[84].
8. Conclusions
NAFLD is considered the chief cause of liver functions abnormality. It is a public health problem since it can progress to NASH, which, in a wide proportion of cases, may lead to endstage liver disease, and may require organ transplantation. Thus,realizing the molecular mechanisms leading to fat accumulation and oxidative balance impairment in steatotic livers is significant.Notably, early detection of NAFLD/NASH, especially in the population at risk, will enable prompt and effective treatment and further management of this multisystem disease. Hence, advances in targeted drug therapy are a priority.
Despite advances in understanding and treating NAFLD, there are still aspects to be resolved, in particular developing early diagnostic markers for the disease. Furthermore, molecular,genetic, and biochemical researches are urgently in need to foster the development of appropriate therapeutic strategies. On the other hand, preclinical animal models are indispensable to explore new drug targets for the development of future therapies. The ideal animal model is the one that closely mimics human disease and reflects correctly both the pathophysiology and histopathology of human NAFLD/NASH. Up until now, no single animal model has included the overall spectrum of human NAFLD progression but possibly will simulate certain characteristics.
From a pharmacological point of view, no decisive pharmacologic therapy has been approved for treatment of NAFLD. All the proposed potential pharmacotherapies are prescribed for NAFLD management including treating the associated obesity, T2DM,hyperlipidemia, and IR.
In the new era in NAFLD management, early intervention and a combination of efficient lifestyle changes and weight loss with potential pharmacological approaches are considered as the cornerstones of NAFLD management.
Conflict of interest statement
The author declares no conflict of interest.
杂志排行

Asian Pacific Journal of Tropical Biomedicine的其它文章
- Aqueous extract of freeze-dried Protaetia brevitarsis larvae promotes osteogenesis by activating β-catenin signaling
- Phloretin-induced suppression of oxidative and nitrosative stress attenuates doxorubicin-induced cardiotoxicity in rats
- Hexadecanoic acid-enriched extract of Halymenia durvillei induces apoptotic and autophagic death of human triple-negative breast cancer cells by upregulating ER stress