2,3,9,10,16,17,23,24-八乙硫基酞菁的合成与理论研究
2022-02-23闫金雪,蔡雪,姜力,马悦
闫金雪,蔡雪,姜力,马悦



摘要:设计合成在酞菁2,3,9,10,16,17,23,24位(β位)八乙硫基取代的分子:2,3,9,10,16,17,23,24-八乙硫基酞菁分子β-(SC2H5)8-H2Pc,并对该分子进行红外吸收光谱表征和理论研究.采用密度泛函理论(DFT)在B3LYP/6-31G(d)水平上对2,3,9,10,16,17,23,24-八乙硫基酞菁分子进行结构优化,模拟其红外吸收光谱.模拟与实验红外振动吸收峰拟合曲线呈线性关系,相关系数R2=0.998,二者符合较好.
关键词:酞菁取代;密度泛函理论;振动光谱;前线分子轨道
[中图分类号]O625[文献标志码]A
Synthesis and Theoretical Study of 2,3,9,10,16,17,23,24-
Octaethylthio Phthalocyanine
YAN Jinxue,CAI Xue*,JIANG Li,MA Yue
(College of Chemistry and Chemical Engineering;Mudanjiang Normal University;Mudanjiang 157011;China)
Abstract:A 2,3,9,10,16,17,23,24-position (β-position) octasubstituted phthalocyanine with ethylthio group,β-(SC2H5)8-H2Pc,was designed and synthesized.This molecule infrared absorption spectrum was characterized,and theoretical research was carried out.Density functional theory (DFT) was used to optimized β-(SC2H5)8-H2Pc at B3LYP/6-31G(d) level, IR spectrum of β-(SC2H5)8-H2Pc was simulated.The simulated and experimental vibration absorption peaks were fitted.The fitted curve shows a linear relationship,the correlation coefficient R2=0.998.The computed results show a good agreement with the tested results.
Key words:phthalocyanine substitution;density functional theory;vibration spectrum;frontier molecular orbital
酞菁(Phthalocyanine,Pc)具有良好的化學热稳定性、低毒性、电学、光学、磁学性质,应用于非线性光学材料、催化剂、光动力疗法中的光敏剂、电化学传感器等领域.[1]酞菁具有丰富的分子修饰位点,周围的16个位点可以被各种类型的取代基取代,产生众多的酞菁衍生物.[23]酞菁作为一种有机功能材料,在其特定位置上引进硫原子可提供一种新型的作用力,以改善溶解度方面的缺陷,有助于后续的加工与应用.本文设计合成β位乙硫基八取代酞菁β-(SC2H5)8-H2Pc,并对该分子进行红外吸收光谱的表征和理论研究,为研究酞菁衍生物性质提供理论依据,为酞菁类化合物的结构和光谱性质的研究提供参考.
1实验
1.1实验合成
4,5-二氯邻二乙硫基苯的合成将4,5-二氯邻苯二甲腈、乙硫醇以及DMF加入到三颈烧瓶中,在氮气的保护下加热到100 ℃,分3次加入无水碳酸钾.磁力搅拌器搅拌,确保四种药品充分混合.在100 ℃的条件下加热7 h,将得到的溶液倒进盛有500 mL冰水的大烧杯中.出现沉淀后,先用循环水真空泵和布氏漏斗抽滤溶液,再依次用水和甲醇洗涤滤渣,获得粗产物.粗产物放入电热恒温鼓风干燥箱中10~15 h,利用柱色谱技术对干燥后的产物进行分离、提纯,得到4,5-二氯邻二乙硫基苯.
β位八乙硫基取代酞菁的合成将正戊醇、4,5-二氯邻二乙硫基苯和金属锂加入到三颈烧瓶中,在氮气的保护下加热回流4 h.待溶液温度降至室温后,加入100 mL甲醇,再将2 mL乙酸滴加到混合溶液中并不断搅拌,使用循环水真空泵和布氏漏斗抽滤溶液,甲醇洗涤滤渣,干燥.以CCl4为淋洗液进行柱层析分离提纯,得到的混合溶液利用旋转蒸发仪去除溶剂,重结晶后得到深绿色产物.
采用NICOLET iS10傅里叶变换红外光谱仪对β-(SC2H5)8-H2Pc分子进行红外吸收光谱(IR)表征测试,波数400~4 000 cm-1.
1.2理论计算采用GaussView 6.0软件建构分子构型.用Gaussian 09程序包[4]在B3LYP/6-31G(d)水平上对β-(SC2H5)8-H2Pc分子的几何结构进行优化,模拟红外吸收光谱.采用GaussView 6.0软件对分子的振动模式进行指认.全部计算过程采用默认的收敛模式,以0.961 4为校正系数对模拟得到的振动光谱频率进行校正.[5]
2结果与讨论
2.1β-(SC2H5)8-H2Pc的结构及优化
对β位八乙硫基取代酞菁分子进行结构优化,得到最稳定结构的β-(SC2H5)8-H2Pc分子.β-(SC2H5)8-H2Pc分子共有114个原子,其中48个C原子,50个H原子,8个N原子,8个S原子.分子结构见图1.
2.2β-(SC2H5)8-H2Pc的红外谱带振动归属
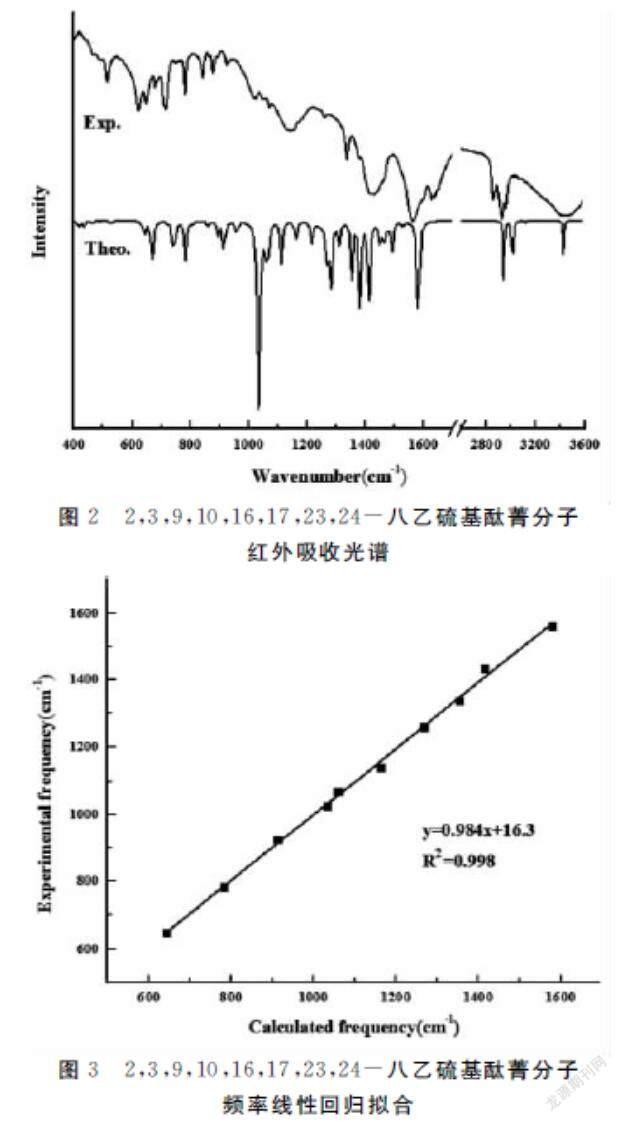
使用GaussView 6.0软件对β-(SC2H5)8-H2Pc分子的振动模式进行指认归属.400~3 600 cm-1范围内的红外吸收光谱图见图2.选取400~2 000 cm-1红外振动吸收峰拟合,模拟与实验红外振动吸收峰拟合曲线呈线性关系,相关系数R2=0.998,理论值与实验值吻合较好,见图3.
红外吸收光谱包含酞菁大环和取代基的典型振动吸收峰.在400~2 000 cm-1范围内多为酞菁特征吸收峰.1 338 cm-1处为酞菁的特征吸收峰,与模拟的红外吸收光谱中1 310 cm-1处相对应.较典型的特征吸收峰在1 425 cm-1和1 564 cm-1处,分别与理论值1 414 cm-1和1 578 cm-1相对应.在2 000 cm-1之后,实验获得的红外吸收光谱中2 925 cm-1对应模拟红外吸收光谱2 937 cm-1,为甲基的特征吸收峰.
模拟红外光谱分析结果表明,644 cm-1归属于酞菁环的苯基对称伸缩振动以及取代基和酞菁外围的C-S伸缩振动;782 cm-1归属于酞菁环的苯基对称伸缩振动;1 033 cm-1归属于酞菁环内C-N不对称伸缩振动;1 111 cm-1归属于酞菁环内C-N对称伸缩和不对称伸缩振动;1 163 cm-1归属于酞菁环内C-N不对称伸缩振动;1 267 cm-1归属于乙基的C-H弯曲振动;1 310 cm-1归属于酞菁大环的伸缩振动;1 414 cm-1归属于酞菁异吲哚伸缩振动;1 578 cm-1归属于异吲哚单元中外围苯基对称伸缩振动;2 937 cm-1归属于甲基的C-H伸缩振动.
2.3β-(SC2H5)8-H2Pc的前线分子轨道
Fukui[6]等认为,分子的许多化学性质通常由前线分子轨道决定.EHOMO值及ELUMO值能够分别体现分子的给电子能力和接受电子能力,EHOMO值越大,越容易失去电子,ELUMO值
越大,则越容易得到电子.通过计算,β-(SC2H5)8-H2Pc分子的轨道能量EHOMO=-4.974 3 eV,ELUMO= -2.943 0 eV,LUMO与HOMO轨道能量差ΔEL-H=2.031 3 eV.由于离域共轭大π键的存在,电子π*→π的跃迁就是HOMO→LUMO的本质.β-(SC2H5)8-H2Pc的前线分子轨道图见图4.
分子的HOMO与LUMO之间的能隙ΔEL-H也可以作为分子活性的判据.依据文献[7]给出的自由酞菁轨道能量EHOMO=-4.976 9 eV,ELUMO=-2.828 7 eV,ΔEL-H=2.148 2 eV,β-(SC2H5)8-H2Pc分子相较于未取代的自由酞菁ΔEL-H更小,故本实验合成的分子具有更好的化学反应活性.
2.4β-(SC2H5)8-H2Pc的分子表面静电势
分子静电势作为分子微观性质中一个重要的物理量,通过对分子表面静电势的分析可以探究分子间的相互作用并确定分子反应的活性位点.β-(SC2H5)8-H2Pc分子的表面静电势分布图由多功能波函数Multiwfn软件[8]和VMD程序[9]得到,见图5,可以精确而直观地展现出分子周围静电作用的分布.取代基周边多为正静电势区域,酞菁的异吲哚单元多为负静电势区域.
分子表面静电势图也是判断分子表面活性位点的一种有效方法.带正电荷的区域容易受到亲核试剂的进攻,存在极大值点80个,最大值点位于与异吲哚苯环中C32相连接的H70附近,最大值为15.78 kcal/mol.带负电荷区域为电子聚集区域亲电试剂更容易向此处进攻,存在极小值点44个,最小值点位于与异吲哚苯环中C27原子相连接的S47附近,最小值为-28.45 kcal/mol(见图1),可以判断出β-(SC2H5)8-H2Pc分子可能参与反应的活性中心,对该分子发生亲电、亲核试剂进攻反应的可能性作出预测.
3结论
设计合成了β位八乙硫基取代酞菁分子,利用密度泛函理论在B3LYP/6-31G(d)水平下对β-(SC2H5)8-H2Pc分子结构进行优化,模拟β-(SC2H5)8-H2Pc分子的红外吸收光谱.理论模拟和实验获得的红外吸收光谱图符合较好,模拟与实验红外振动吸收峰的拟合曲线呈线性关系,相关系数R2=0.998,说明所采用的计算方法适用于酞菁分子的理论研究.采用GaussView 6.0软件对β-(SC2H5)8-H2Pc分子的简正振动模式进行指认归属;分析了β-(SC2H5)8-H2Pc的前线分子轨道,计算出该分子的HOMO-LUMO能级差,并对该分子的分子静电势及其极大值与极小值进行了讨论.研究成果可为酞菁衍生物性质的研究提供理论依据,为酞菁类化合物的结构和光谱性质研究提供参考.参考文献
[1]Riccardis A D,Lee M,Kazantsev R V,et al.Heterogenized pyridinesubstituted cobalt(II) phthalocyanine yields reduction of CO2 by tuning the electron affinity of the Co center[J].ACS Applied Materials & Interfaces,2020,12(5):52515258.
[2]赵玥,周启.甲氧基外围取代四苯并氮杂卟啉结构与性质研究[J].牡丹江师范学院学报:自然科学版,2021(3):3034.
[3]李梓萌,蔡雪.咔唑类取代酞菁锌染料敏化性能研究[J].牡丹江师范学院学报:自然科学版,2016(1):4446.
[4]Frisch M, Trucks G,Schlegel H,et al.Gaussian 09 (Revision D.01)[CP].Wallingford CT:Gaussian,Incorporated,2009.
[5]Alecu I M,Zheng J,Yan Z,et al.Computational Thermochemistry:Scale Factor Databases and Scale Factors for Vibrational Frequencies Obtained from Electronic Model Chemistries[J].Journal of Chemical Theory & Computation,2010,6(9):28722887.
[6]Fukui K,Koga N,Fujimoto H.Interaction frontier orbitals[J].Journal of the American Chemical Society,2015,103(1):196197.
[7]蔡雪.在密度泛函理論基础上研究酞菁类化合物的结构,光谱及OFET性质[D].济南:山东大学,2009.
[8]Lu T,Chen W.Multiwfn:a multifunctional wavefunction analyzer[J].Journal of Computational Chemistry,2012,33:580592.
[9]Humphrey W,Dalke A,Schulten K.VMD:Visual molecular dynamics[J].Journal of Molecular Graphics and Modelling,1996,14(1):3338.
编辑:吴楠
