中年男性,进行性行走不稳9年
——成人晚发型常染色体隐性遗传小脑性共济失调1型
2022-01-08郑浩然曹立田沃土
郑浩然 曹立 田沃土
1 临床资料
患者,男,44岁,因“进行性行走不稳9年”就诊,患者自35岁起出现行走速度慢,易摔倒,并伴有言语含糊不清,旁人难以理解,病情随时间逐渐加重。六年前,患者出现书写笨拙;五年前,开始出现进食流食后频繁呕吐。现上下楼梯需搀扶,否认长期服用和接触酒精或药物史,否认感染和卒中史等。患者出生自近亲婚配家庭,父母为两代以内堂兄妹,家族中无相似共济失调症状(图1 A)。
体格检查:一般检查及心、胸、腹部查体未见异常,神志清,吐词欠流利,眼震轻度,视力粗测正常,四肢肌力5级;腱反射:双上肢(+),双下肢(++);病理征(-),无高弓足,感觉正常,双侧指鼻试验、跟膝胫试验和轮替试验均欠稳准(表1)。患者时隔一年来我院进行首诊和随访时,分别使用国际协作共济失调评估量表(International collaborative ataxia assessment scale,ICARS)进行评估结果如下:一年前后肢体共济失调(15/52 vs.15/52)、言语障碍(3/8 vs.3/8)和动眼功能(1/6 vs.1/6)均未见明显恶化,而姿势和步态障碍(3/34 vs.5/34)略有进展(表 2)。

表1 ARCA-1患者的临床特征

表2 ARCA-1患者共济失调量表(ICARS)
辅助检查:血、尿、便常规正常,肝肾功能、维生素浓度等均正常。眼底镜检查正常。头颅MRI显示中度弥漫性小脑萎缩,其余未见明显异常(图1 B)。
遗传学检查:运用遗传性共济失调Panel进行二代测序(测序基因总数:182,测序深度:200X,覆盖度:大于98%)结果提示患者(IV:1)存在 SYNE1基因(NM_182961)纯合移码突变 c.9347delA(p.N3116Mfs*6),家系共分离和一代测序验证提示父亲(III:2)为相同突变的杂合携带者(母亲因已去世未获得标本进行基因检测)。根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)评估和分级指南[1],该变异被评为致病变异(PVS1+PM2+PP3)(图1 A、C)。
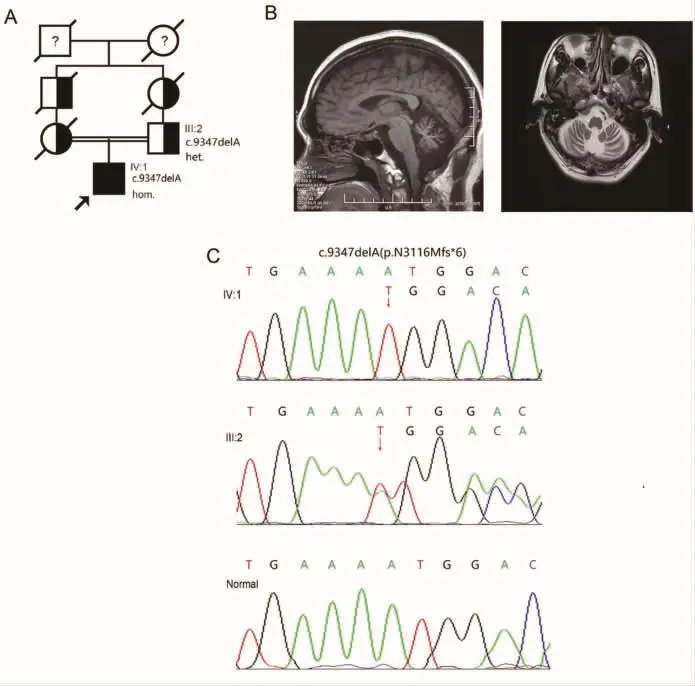
图1 ARCA-1患者家系图,影像学检查及基因一代测序结果A.家系图示患者出生自近亲婚配家庭,父母为两代以内堂兄妹。B.患者头颅MRI显示中度弥漫性小脑萎缩。C.患者(IV:1)存在SYNE1 基因(NM_182961)纯合移码突变 c.9347delA(p.N3116Mfs*6),父亲(III:2)为该移码突变杂合携带者。
本例患者表现为晚发性、缓慢进展的的单纯型小脑性共济失调并伴有构音障碍,其来自近亲婚配家庭,结合其临床表现、辅助检查结果以及基因检测结果,综合考虑其诊断符合常染色体隐性遗传小脑性共济失调1型(autosomal recessive cerebellar ataxia type 1,ARCA-1)。
2 讨论
ARCA-1是一种呈常染色体隐性遗传的小脑性共济失调,表现为晚发、进展缓慢、单纯型共济失调,并伴有构音障碍。本病最初是在一群来自魁北克东南部的布尔斯地区的法裔加拿大家庭中被发现的[2]。到目前为止,全世界有超过90例ARCA-1患者报告,大多数患者为法裔加拿大人。ARCA-1在白种人中患病率较高。在排除弗里德赖克共济失调和最常见的重复序列扩增引起的脊髓小脑性共济失调(spinocerebellar ataxias,SCAs)后,对 434名白种人患者进行SYNE1突变筛查,约5%诊断为ARCA-1[3]。此外,在巴西、日本、土耳其、摩洛哥、阿尔及利亚和斯里兰卡等地区报告了约10例患者[3-7]。除了小脑共济失调和构音障碍外,在以往病例中还有其他表型被报告,如辩距不良、下肢腱反射活跃、扫视和追踪异常等;影像学检查可发现弥漫性单纯性小脑萎缩[2,7]。既往,ARCA-1被认为是一种临床表现一致性较高的神经退行性疾病[2]。然而,最近一项对26例ARCA-1患者的研究显示,其中21例除具有典型ARCA-1临床表现外,还可合并其他表型,如肌萎缩性脊髓侧索硬化症(amyotrophic lateral sclerosis,ALS)表现、周围神经病变、精神发育迟滞、脑干功能障碍和肌肉骨骼异常[3]。此外,来自近亲婚配家庭的两例日本患者和两例土耳其患者曾被诊断为运动神经元疾病伴或不伴脊髓小脑共济失调[4,6]。这些发现表明ARCA-1具有一定的临床异质性。与其他大多数遗传性共济失调疾病相比,ARCA-1型的共济失调进展相对缓慢,且致残程度也相对较轻[2]。对64例遗传学诊断为ARCA-1的患者进行分析,共济失调平均发病年龄为31.6±7.8岁 (17~46岁),其主要临床症状的严重程度处于轻至中度 (评估时患者平均病程为14.33年)[2]。2019年,中国大陆另一有关ARCA-1的临床病例报告中3例ARCA-1先证者平均发病年龄为22.3岁(18~25岁),3例分别在相应病程的第8年、第4年和第10年时进行ICARS评估,得分分别为32、41和42分,除具有ARCA-1的典型临床特点外,其中的两名患者还伴有上运动神经元损害(痉挛步态、下肢腱反射活跃、病理征阳性)[8];而本文先证者在起病后第9年的ICARS评分较上述3例患者低,发病年龄也相对较晚,不具有上运动神经元损害的体征;考虑可能与不同突变对SYNE1蛋白功能影响不同以及ARCA-1的表型异质性有关。
SYNE1是ARCA-1的致病基因,迄今,世界上已报告了大约50种突变,包括无义突变、错义突变、移码突变和剪接位点突变[2-3,6-7]。其中大多数突变被预测导致相应编码蛋白发生截短。在124例法裔加拿大ARCA-1患者中,具有内含子突变g.306434A>G的患者占总数50.8%,这可能与奠基者效应有关[2]。到目前为止,在ARCA-1中尚未发现基因型-表型相关性。移码突变在SYNE1的突变谱中相对少见[2]。SYNE1基因共有146个外显子,本研究所报告的新致病突变 c.9347delA(p.N3116Mfs*6)为位于第 59号外显子的移码突变,其可导致编码一错误截短型蛋白,后者丢失64.5%的正常氨基酸序列。因此,该移码突变很可能通过无义介导的mRNA降解机制(nonsense-mediated mRNA decay,NMD)导致疾病的发生[9]。
SYNE1位于染色体6q25.2上,是人类基因组中最大的基因之一。它的产物为一含有8797个氨基酸的Nesprin1 giant蛋白,在小脑皮层的浦肯野氏细胞和脑干橄榄区神经元中大量表达[2]。SYNE1是连接质膜和肌动蛋白细胞骨架的结构蛋白spectrin家族成员[10]。spectrin家族还包括PLEKHG4[11]和 SPTBN2[12],它们分别是 16q相关的常染色体显性遗传性小脑性共济失调和脊髓小脑性共济失调5型的致病基因。有趣的是,两者均具有晚发性、缓慢进展性、单纯性小脑共济失调等特点,表现出与ARCA-1的相似之处。因此,ARCA-1与上述两种疾病一样,属于结构蛋白spectrin家族相关的遗传性共济失调[2]。然而,为何这三种蛋白的突变会导致类似的临床表现,其机制仍然未知。Nesprin 1 giant含有 c-末端 Klarsicht/Anc1/Syne1同源性(KASH)域,在ARCA-1的发病机制中起关键作用;研究表明,KLNes1g(为具有 KASH-LESS Nesprin1 giant变异蛋白)在小脑中表达最丰富,提示ARCA-1靶向影响小脑功能[13]。另一项动物研究表明,不影响KASH域的SYNE1 KO小鼠没有发病,而影响KASH域的两只SYNE1 KO小鼠表现出Emery Dreifuss肌营养不良样表型[14]。所有已报告SYNE1突变(包括本文),均位于KASH域的上游,提示在疾病的发展过程中,KASH域缺失的SYNE1可能发挥了其特有的致病性作用。目前 ARCA-1的临床与发病机制研究仍十分有限,随着高通量测序技术的广泛应用,将有助于该类患者得到及时的遗传学诊断;进一步探讨SYNE1致病性突变所引起的分子生物学功能改变,将为本病的诊治提供更多线索。
3 点评
SYNE1基因突变所致ARCA-1常于成人期起病,其临床以共济失调为主要特点,病程进展相对较慢。目前,国内临床医生对该疾病的认识仍然十分有限,对于发病较晚、症状相对单一的成人患者,需进一步行ARCA相关的基因检测,将有助于本病的临床和遗传学诊断。
