马来酸酐改性茶多酚对非酶糖基化的抑制作用
2022-01-06代杨艳韩宇琴廖兵武黄惠华
代杨艳,韩宇琴,廖兵武,廖 晶,黄惠华
(华南理工大学食品科学与工程学院,广东 广州 510641)
非酶糖基化反应又称美拉德反应,是一种广泛存在于食品生产过程中的褐变反应。还原糖与蛋白质或脂质通过非酶反应结合形成不可逆的非酶晚期糖基化终产物(advanced glycosylation end products,AGEs)[1]。虽然美拉德反应的发生对于一些食品品质有正面作用(例如有助于面包的色泽、香气风味等的形成),但是在食品加工过程中的美拉德反应往往涉及高温处理(如烘焙、烹饪、乳制品和饮料生产等),会导致营养物质的损耗和有害成分的产生;同时有研究表明,AGEs在人体中的积累与许多慢性退行性疾病如糖尿病[2]和肾脏疾病[3]的产生有着密切的联系,此外AGEs也是导致肿瘤形成和病变的原因之一[4]。研究者发现通过减少中间产物的生成,阻隔氧化过程以及阻断晚期糖基化终产物受体可以抑制AGEs的生物合成。但是人工合成的抑制剂氨基胍(amino guanidine,AG)对人体具有一定的毒副作用,因此,寻找高效低毒的天然抑制剂成为研究热点。
我国是茶叶生产大国,茶叶资源十分丰富,茶叶中多酚类物质含量依茶叶种类不同而有差别,约占茶叶干基质量的18%~30%左右,其中儿茶素是茶多酚(tea polyphenols,TP)的主要成分,约占茶叶中多酚总量的70%~80%[5]。近年来许多研究发现茶多酚具有显著的抗氧化[6]、抗肿瘤[7]、抑菌抗炎[8]、降血脂[9]、防治心脑血管疾病[10]、提高免疫能力[11-12]、预防和治疗糖尿病[13]等活性功能。此外,研究表明类黄酮[14]、酚酸[15]、芪类[16]对非酶糖基化具有抑制活性。多酚类化合物的生物学功能主要取决于其溶解性和稳定性,TP可溶于水,但在水中易受光或氧化等因素降解并发生褐变[17]。对TP进行化学改性,将亲水性取代基修饰到表没食子儿茶素没食子酸酯(epigallocatechin gallate,EGCG)等儿茶素的分子结构中,从而改变TP的物理化学性质,增强溶解性和稳定性并延缓其氧化,同时能够保留或增强TP的功能活性。Moon等[18]采用酶催化法合成3 种表没食子儿茶素没食子酸苷,结果表明3 种表没食子儿茶素没食子酸苷的抗氧化能力略有下降,但其水溶性大幅提高,同时光稳定性和抗褐变能力也得到提升。马来酸酐(maleic anhydride,MA)是目前世界上仅次于苯酐和醋酐的第三大酸酐原料,由于其含有共轭马来酰基,水解后具有很强的亲水性,化学性质非常活泼,易发生酰胺化反应和酯化反应,因此可以用于某些天然成分的改性。基于此,本研究采用MA对TP进行化学改性,探究改性TP的结构、抗氧化活性及抗糖化能力,以期为改性TP作为非酶糖基化抑制剂在食品领域提供理论参考。
1 材料与方法
1.1 材料与试剂
TP、EGCG、儿茶素(catechin,C)、表儿茶素(epicatechin,EC)、表没食子儿茶素(epigallocatechin,EGC)、表儿茶素没食子酸酯(epicatechin gallate,ECG)、咖啡因(caffeine,CAF)标准品(纯度98%) 上海源叶生物科技有限公司;MA天津市科密欧化学试剂有限公司;6-羟基-2,5,7,8-四甲基色烷-2-羧酸(6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid,Trolox) 美国Sigma公司;喹喔啉、2-甲基喹喔啉、2-(2,3,4-三羟基丁基)-喹喔啉标准品北京百灵威公司;乙腈(色谱纯) 永华化学科技(江苏)有限公司;赖氨酸(lysine,Lys)、果糖(fructose,Fru)、AG、吡啶、重水(D2O)、2,2’-联氮-双-3-乙基苯并噻唑啉-6-磺酸(2,2’-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid,ABTS)、邻苯二胺(o-phenylenediamine,OPD) 上海麦克林生化科技有限公司。
1.2 仪器与设备
UltiMate 3000高效液相色谱(high performance liquid chromatography,HPLC)仪 美国DIONEX公司;Synergy LX多功能微孔板检测仪 美国BioTek仪器有限公司;AVANCE 400MHz液体超导核磁共振(nuclear magnetic resonance,NMR)波谱仪、VERTEX-33傅里叶变换红外光谱(Fourier transform infrared spectroscopy,FTIR)仪 德国Bruker公司;SHA-B恒温振荡器 常州澳华仪器有限公司;HH-4恒温数显水浴锅 常州普天仪器制造有限公司;RE-52AA旋转蒸发器 上海亚荣生化仪器厂;LGJ-10冷冻干燥机 北京松源华兴科技发展有限公司;UV-1800紫外-可见分光光度计 日本岛津公司;PHS-25 pH计 上海仪电科学仪器股份有限公司。
1.3 方法
1.3.1 马来酸酐改性茶多酚的制备
马来酸酐改性茶多酚(maleic anhydride modified tea polyphenols,MA-TP)的制备根据白艳等[19]的方法并加以改动。准确称取TP样品0.875 g于50 mL乙酸乙酯中,加入0.786 g MA和0.5 mL吡啶,置于50 ℃恒温水浴摇床反应6 h。所得样液通过低温旋蒸后冷冻干燥,得到MA-TP粉末。按照相同的方法制备MA改性EGCG(maleic anhydride modified EGCG,MA-EGCG)粉末。
1.3.2 结构表征
1.3.2.1 茶多酚组分鉴定
根据胡湘蜀[20]、Liang[21]等的方法并适当改动鉴定TP组分。采用HPLC鉴定TP样品主要成分,准确称取0.1 g TP样品溶于乙腈中,用棕色容量瓶定容至100 mL,制备得到1 mg/mL的TP溶液,用0.22 μm滤膜过滤。色谱条件:色谱柱:Eclipse XDS-C18(250 mm×4.6 mm,5 μm);流动相:A相为体积分数0.1%甲酸,B相为乙腈,流动相使用前用0.45 μm滤膜过滤;梯度洗脱程序:0~5 min,95%~80%流动相A,25~40 min,80%~50%流动相A,40~45 min,50%~90%流动相A,45~50 min,90%~95%流动相A;进样量:10 μL;流速:1 mL/min;柱温:30 ℃;紫外检测器检测波长280 nm。
1.3.2.2 傅里叶变换红外光谱分析
取少量干燥TP、MA-TP和TP/MA(1∶1,m/m)物理混合样品,与适量KBr粉末均匀混合,压片制样,放置在FTIR仪中进行检测。测定条件:分辨率4 cm-1,扫描范围4 000~400 cm-1。
1.3.2.3 核磁共振分析
将适量的EGCG和MA-EGCG溶解于重水(D2O)中(终质量浓度40 mg/mL),采用400 MHz扫描,测定各化合物的1H-NMR和13C-NMR。
1.3.3 抗氧化活性测定
根据Re等[22]的方法适当改动。将7 mmol/L ABTS与2.45 mmol/L过硫酸钾溶液混合,避光室温放置16 h,配制成ABTS阳离子自由基溶液,用pH 7.4磷酸盐缓冲液(phosphate buffered saline,PBS)将ABTS阳离子自由基溶液稀释直至734 nm波长处吸光度为0.70±0.02。吸取适量样品(TP、MA-TP),加入ABTS阳离子自由基溶液3 mL混合使其终质量浓度分别为20、40、60、80、100 μg/mL,室温下避光反应4 min,在734 nm波长处测定吸光度A样品。以VC作为阳性对照。实验重复3 次,按式(1)计算ABTS阳离子自由基清除率。通过线性拟合方法计算各组样品的半抑制浓度(median inhibition concentration,IC50)。以Trolox为标准品(0.2、0.4、0.6、0.8、1.0、1.2 mmol/L)绘制Trolox浓度-ABTS阳离子自由基清除率标准曲线,根据标准曲线方程计算各样品ABTS阳离子自由基清除能力,结果以每克样品清除ABTS阳离子自由基相当于Trolox的物质的量表示,单位为mmol/g。
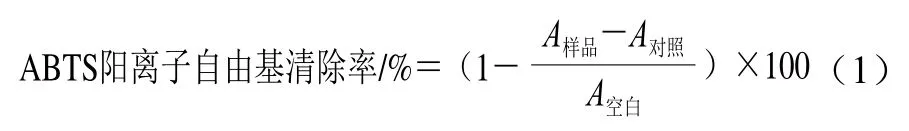
式中:A样品为样品+ABTS阳离子自由基溶液的吸光度;A对照为样品+PBS的吸光度;A空白为PBS+ABTS阳离子自由基溶液的吸光度。
1.3.4 非酶糖基化模拟体系的构建
根据Ajandouz等[23]的方法适当改动构建非酶糖基化模拟体系。用0.2 mol/L pH 7.4 PBS分别配制0.05 mol/L赖氨酸溶液和0.05 mol/L果糖溶液,用PBS配制质量浓度为0.3 mg/mL的TP、MA-TP样品溶液,以AG为阳性对照。按照表1建立4个非酶糖基化模拟体系,分别为样品抑制体系Fs、不含果糖对照体系Fa、完全糖基化对照体系Fb、不加样品和不含果糖对照体系Fc。各组反应液在90 ℃油浴中反应5 h,分别于反应0、0.5、1、2、3、4、5 h时迅速取出各组反应液并进行冰水浴冷却,置于-20 ℃冰箱保存待测。

表1 非酶糖基化模拟体系构成Table 1 Composition of simulated non-enzymatic glycosylation systems
1.3.5 非酶糖基化反应产物分析
取1 mL不同加热反应时间的各糖基化模拟体系反应液,用0.2 mol/L pH 7.4 PBS稀释10 倍后,分别于294、420 nm波长处测吸光度,空白对照为PBS。通过吸光度反映体系中非酶糖基化产物生成情况,比较不同样品对非酶糖基化反应的抑制能力。
1.3.6 二羰基化合物抑制率的测定
二羰基化合物浓度的测定根据Kocadagh等的方法[24]适当改动。配制一系列的喹喔啉、2-甲基喹喔啉和2-(2,3,4-三羟基丁基)-喹喔啉的标准溶液(0.01、0.025、0.05、0.1、0.25、0.5 mmol/L),过0.22 μm有机滤膜后进行HPLC分析,获得计算乙二醛(glyoxal,GO)、甲基乙二醛(methylglyoxal,MGO)、3-脱氧葡萄糖醛酮(3-deoxyglucosone,3-DG)浓度的标准曲线方程。用0.2 mol/L pH 7.4 PBS配制0.05 mmol/L的OPD溶液,取1 mL 1.3.4节不同加热反应时间的各组糖基化反应液样品,加入300 μL OPD溶液,在60 ℃水浴中衍生30 min,过0.22 μm有机滤膜后进行HPLC分析。色谱条件:色谱柱:Eclipse XDS-C18(250 mm×4.6 mm,5 μm);流动相:A相为体积分数0.1%甲酸,B相为甲醇,流动相使用前用0.45 μm滤膜过滤;梯度洗脱条件:0~10 min,A相70%~40%;10~12 min,A相40%~70%;A相70%持续3 min,运行总时间为15 min;进样量:5 μL;流速:1 mL/min;柱温:30 ℃;紫外检测器检测波长315 nm。通过标准曲线方程计算二羰基化合物浓度;按式(2)计算各二羰基化合物抑制率。
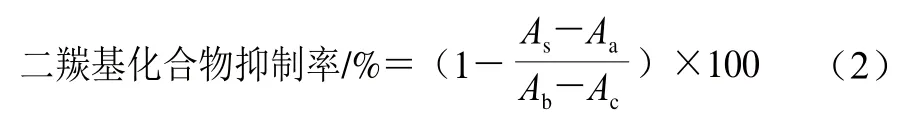
式中:As为样品+Lys+Fru体系中各二羰基化合物峰面积;Aa为样品+Lys体系中各二羰基化合物峰面积;Ab为Lys+Fru体系中各二羰基化合物峰面积;Ac为只含Lys体系各二羰基化合物峰面积。
1.3.7 戊糖素抑制率的测定
根据Awasthi等[25]的方法适当改动测定戊糖素含量。取1 mL 1.3.4节不同加热反应时间的各组糖基化反应液,用PBS稀释30 倍。于激发电压600 V、激发波长335 nm、发射波长382 nm条件下测定反应液荧光强度,空白对照为PBS,以荧光强度表征戊糖素含量。按式(3)计算戊糖素抑制率。
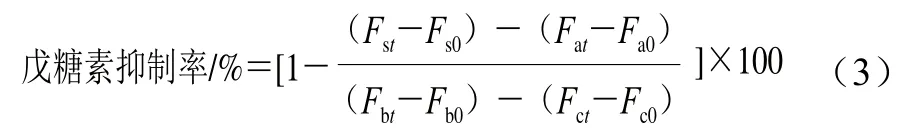
式中:Fst和Fs0为样品+Lys+Fru体系分别在t时刻和0时刻的荧光强度;Fat和Fa0为样品+Lys体系分别在t时刻和0时刻的荧光强度;Fbt和Fb0为Lys+Fru体系分别在t时刻和0时刻的荧光强度;Fct和Fc0为只含Lys体系分别在t时刻和0时刻的荧光强度。
1.3.8 荧光性AGEs抑制率的测定
根据Awasthi等[25]的方法适当改动。取1 mL 1.3.4节不同加热反应时间的各组糖基化反应液,用PBS稀释30 倍。于激发电压600 V、激发波长370 nm、发射波长440 nm条件下测定荧光强度,空白对照为PBS,以荧光强度表征荧光性AGEs含量。按式(4)计算荧光性AGEs抑制率。
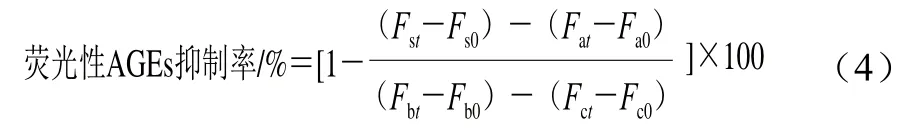
式中:Fst和Fs0为样品+Lys+Fru体系分别在t时刻和0时刻的荧光强度;Fat和Fa0为样品+Lys体系分别在t时刻和0时刻的荧光强度;Fbt和Fb0为Lys+Fru体系分别在t时刻和0时刻的荧光强度;Fct和Fc0为只含Lys体系分别在t时刻和0时刻的荧光强度。
1.4 数据处理与分析
实验进行3 组平行,3 次重复,所有数据采用Origin Pro 8.5软件进行处理,结果以平均值±标准差表示。使用SPSS 19.0统计分析软件对数据进行单因素方差分析和Duncan检验,分析差异显著性,以P<0.05表示差异显著。
2 结果与分析
2.1 茶多酚结构表征
2.1.1 茶多酚组分分析结果
由图1可知,TP各组分得到很好的分离,主要成分为EGCG、ECG、C、EC、EGC等儿茶素单体以及CAF。与儿茶素单体及CAF标准品图谱对比,得到TP样品各组分的相对含量分别为EGCG 66.20%、ECG 22.06%、EC 3.91%、EGC 2.31%、CAF 1.16%、C 0.44%。
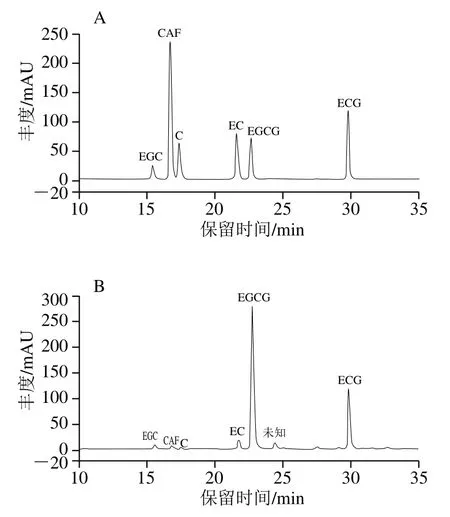
图1 5 种儿茶素单体和CAF标准品(A)与TP样品(B)的HPLC图Fig. 1 HPLC chromatograms of mixed standard solution of five catechin monomers and CAF (A) and TP sample (B)
2.1.2 傅里叶变换红外光谱分析结果
TP、MA-TP和TP/MA物理混合样品的FTIR图如图2所示。3 444 cm-1处吸收峰是苯环上Ar—OH的伸缩振动所引起;1 692 cm-1处吸收峰是羰基C=O伸缩振动所引起;1 613、1 519、1 453 cm-1处吸收峰是苯环面内C=C骨架振动所引起;1 340 cm-1处吸收峰是苯环上O—H的面内弯曲振动所引起;1 236 cm-1处吸收峰是C—O—C的反对称伸缩振动所引起;1 033 cm-1处吸收峰是C—O—C对称伸缩振动所引起;969 cm-1处吸收峰是C=C的反式伸缩振动所引起[26-28]。

图2 TP、MA-TP、TP/MA物理混合的FTIR图Fig. 2 Fourier transform infrared spectra of TP, MA-TP and TP/MA physical mixture
C—O—C的反对称吸收峰和对称吸收峰通常分别出现在1 270~1 230 cm-1和1 050~1 000 cm-1处,MA-TP在1 236 cm-1和1 033 cm-1处吸收峰强度均强于TP和TP/MA,在1 694 cm-1处具有羰基C=O伸缩振动的强吸收峰,表明改性后的产物属于一种酯类物质[29];同时,MA-TP在970 cm-1处吸收峰强度明显强于TP,可能是MA结构中原有的C=C结合到TP分子中所引起[30];另一方面,在TP/MA中发现了1 848 cm-1处和1 776 cm-1处的吸收峰,分别为MA重复单元中C=O的对称和反对称伸缩振动吸收峰[31],而在MA-TP图谱中没有出现,说明MA已基本上参与了反应且无残留。
2.1.3 核磁共振光谱分析结果
由于TP是一种混合物,不易对其进行核磁共振光谱分析,同时上述液相分析结果表明EGCG是TP样品中含量最高的儿茶素,所以通过相同的改性方法对EGCG进行改性得到MA-EGCG粉末,并对EGCG和MA-EGCG进行核磁共振光谱分析来检测MA改性对EGCG以及TP结构的影响。由图3A1、B1中1H-NMR图谱可知,MA-EGCG各质子峰的化学位移与EGCG相近,但与EGCG相比,MA-EGCG 8位C上化学位移δ=5.92的质子峰[32]消失,同时在化学位移δ=6.17、δ=7.83、δ=8.37、δ=8.55处新增了较强的质子峰,表明MA改性可能取代了8位C上的H,并根据新增质子峰推测可能发生了多取代反应,后续将通过质谱进行进一步结构鉴定。由图3A2、B2中13C-NMR图谱可知,MA-EGCG的化学位移与EGCG相近,新增了δ=127.30、δ=133.60、δ=138.13、δ=170.55处4个较强的峰,可能为MA结构中C=C、酯键中碳的化学位移[33]。同样,在MA-TP的1H-NMR和13C-NMR的图谱中,也有相应的化学位移出现(文中未给出图)。

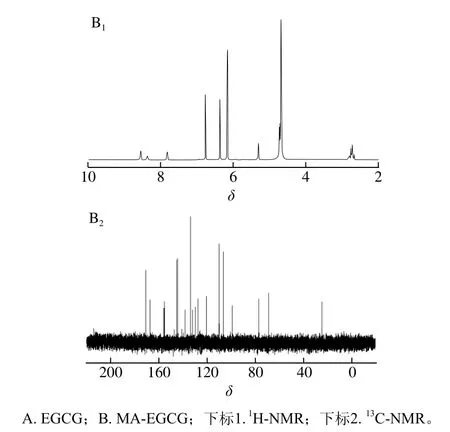
图3 EGCG和MA-EGCG的1H-NMR和13C-NMR图Fig. 3 1H-nuclear magnetic resonance (NMR) and 13C-NMR spectra of EGCG and MA-EGCG
2.2 茶多酚改性前后抗氧化活性比较
如图4所示,在质量浓度为20~100 μg/mL范围内,TP和MA-TP比VC具有更高的ABTS阳离子自由基清除率,说明TP和MA-TP均具有良好的抗氧化活性。但是当质量浓度低于100 μg/mL时,MA-TP的ABTS阳离子自由基清除率显著低于TP(P<0.05),这可能是由于MA化学改性使TP分子结构上的部分活性羟基被取代,从而导致抗氧化活性下降。经计算,TP、MA-TP和VC ABTS阳离子自由基清除率的IC50分别为(22.19±0.15)、(43.49±1.06)、(76.98±0.10)μg/mL;而这3 种样品的半抑制浓度下ABTS阳离子自由基清除能力分别为(15.86±0.11)、(8.09±0.03)、(4.57±0.01)mmol/g。表明在本实验浓度范围内,3 种物质对ABTS阳离子自由基清除能力的顺序由高到低为TP>MA-TP>VC。

图4 TP、MA-TP和VC对ABTS阳离子自由基的清除作用Fig. 4 ABTS cation radical scavenging activity of TP, MA-TP and VC
2.3 改性茶多酚对非酶糖基化产物的影响
非酶糖基化产物在294 nm波长处的吸光度是反映体系中前体物质形成情况的特征指标[23],因此本实验测定了不同加热时间下的不同反应组别(Lys+Fru完全糖基化组、AG+Lys+Fru阳性对照组、TP+Lys+Fru及MTP+Lys+Fru抑制组)在294 nm波长处的吸光度,结果如图5A所示,各组别吸光度都随着加热反应时间的延长而明显上升,这与Kim等[34]对葡萄糖-甘氨酸/二甘氨酸/三甘氨酸体系进行热处理的研究结果一致。但在相同加热反应时间内,各组A294nm由低到高依次为MA-TP+Lys+Fru<TP+Lys+Fru<AG+Lys+Fru<Lys+Fru。结果表明,随着加热反应时间的延长,大量前体物质生成;同时,MA-TP和TP具有优于阳性对照的前体物质抑制效果,并且MA改性提高了TP对前体物质的抑制能力,MA-TP对中间产物的较强抑制效果可能是其结构对前体物质的捕获能力增强所致。
非酶糖基化反应的高级阶段会生成棕色物质,通常以其在420 nm波长处的吸光度来反映非酶糖基化程度[35]。由图5B可知,不同反应组A420nm随着加热反应时间的延长明显增大,而在相同加热时间下,各组A420nm由低到高依次为MA-TP+Lys+Fru<TP+Lys+Fru<AG+Lys+Fru<Lys+Fru。上述结果表明,随着加热时间的延长,棕色物质快速生成并积累,与上述前体物质有相似的增长趋势,Benjakul等[36]的研究也发现反应体系中生成的前体物质促进了焦糖化反应的发生;同时通过与阳性对照AG相比,MA-TP和TP具有更好的抑制效果,且MA改性提高了TP对棕色物质的抑制能力。
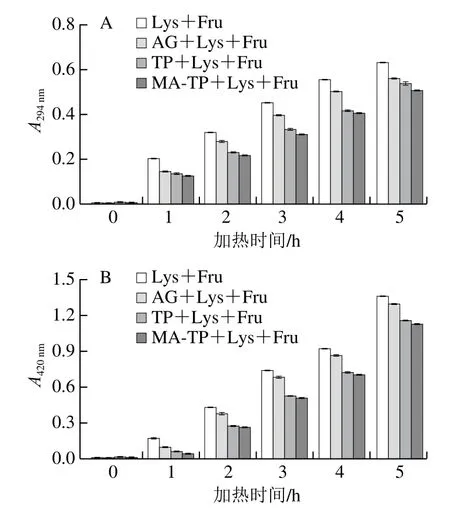
图5 果糖-赖氨酸模拟体系中得到的非酶糖基化反应产物在294 nm(A)、420 nm(B)波长处的吸光度随加热时间的变化Fig. 5 Changes in absorbance at 294 nm (A) and 420 nm (B) of the non-enzymatic glycosylated products derived from aqueous fructoselysine model system as a function of heating time
2.4 茶多酚及其改性衍生物对二羰基化合物的抑制作用
在非酶糖基化反应的中间阶段,Amadori产物经过一系列脱氢、氧化和重排反应生成具有高度活性的衍生羰基化合物,其中GO、MGO和3-DG是人体内AGEs的重要中间体[37]。图6为3 种α-二羰基化合物标准品及模拟体系反应液经过OPD衍生化后在315 nm波长处的HPLC图,可以看出模拟体系反应液中3 种α-二羰基化合物GO、MGO、3-DG得到了较好的分离。3 种α-二羰基化合物GO、MGO、3-DG的标准曲线方程分别为y=6.360 1x+0.015 5、y=8.555 7x+0.016 9、y=11.031 9x+0.001 2,决定系数R2均大于0.999 8,表明3 种化合物标准曲线的线性关系良好。
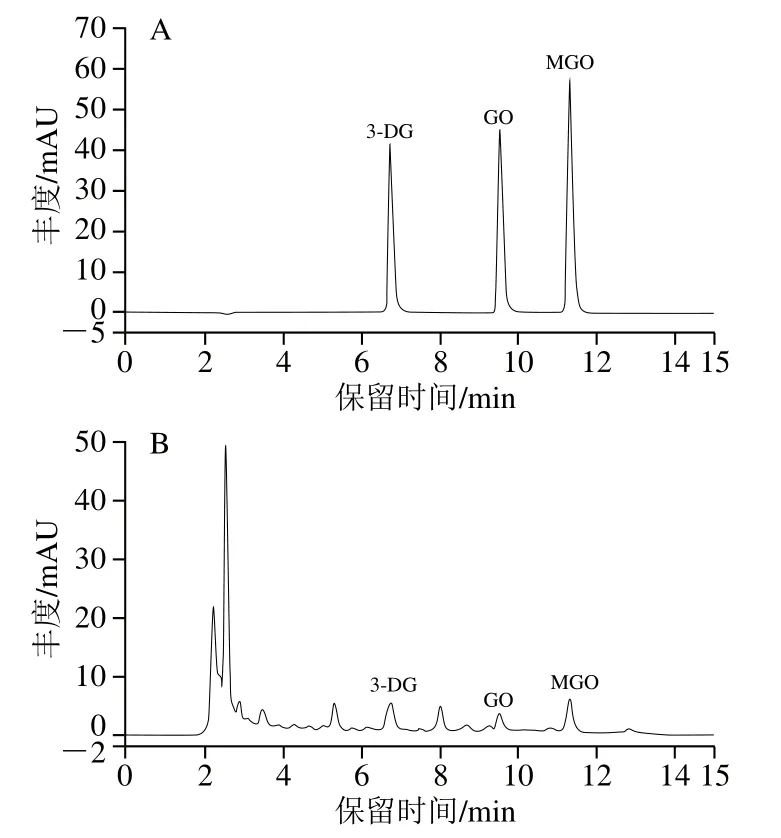
图6 3 种二羰基化合物标准溶液(A)和反应体系样液(B)的HPLC图Fig. 6 HPLC chromatograms of three α-dicarbonyl standards (A) and Maillard reaction solution (B)
图7A为GO浓度随加热时间的变化,在加热过程中GO浓度开始阶段迅速升高,随后下降,变化趋势与黄启瑞等[38]的研究结果相似,这可能是由于在反应初期GO快速生成,进入非酶糖基化反应末期阶段其被消耗。完全糖基化组和阳性对照组的GO浓度在加热2 h达到最大,而TP和MA-TP抑制组在加热3 h达到最大,这可能是由于TP和MA-TP具有捕获GO的能力[39],使反应前期GO生成速度减慢,而随着加热时间的延长抑制剂被消耗,后期难以抑制GO的生成。在相同的加热时间下,MA-TP抑制组中的GO浓度始终显著低于TP抑制组(P<0.05),抑制率最高为82.95%(0.5 h时),表明MA-TP对GO的抑制能力强于TP,说明经过MA改性后,MA-TP的化学结构更有利于对GO的捕获。图7B为MGO浓度随加热时间的变化,各组MGO浓度随加热时间的延长而增加,这与吕梦莎等[40]的研究结果相似,但在相同的加热时间下,MA-TP、TP抑制组和阳性对照组中MGO浓度显著低于完全糖基化处理组,表明MA-TP、TP和AG对MGO的生成均具有良好的抑制效果;同时MA-TP抑制组中MGO浓度始终显著低于TP组(P<0.05),抑制率最高为40.33%(3 h时),表明MA-TP对MGO的抑制能力也强于TP,同样说明经过MA改性后,MA-TP更有利于对MGO的捕获;图7C为3-DG浓度随加热时间的变化,各组3-DG浓度随加热时间的延长而增加,这与吴春剑[41]的结果相似。MA-TP、TP和AG对3-DG的生成同样均具有良好的抑制效果。反应前期MA-TP、TP抑制组中3-DG浓度相近,这可能是由于3-DG分子结构大,经过MA改性后的MA-TP的空间位阻大,不易与3-DG结合,MA-TP抑制率最高为45.01%(5 h时)。但在反应中后期,MA-TP抑制组中3-DG浓度显著低于TP(P<0.05),这可能是由于经过MA改性,TP热稳定性增强,使得MA-TP在反应中后期对3-DG的生成依旧具有良好的抑制能力。
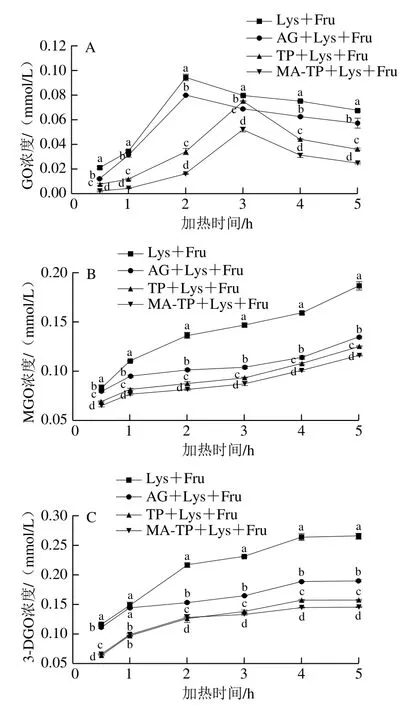
图7 果糖-赖氨酸反应体系中GO(A)、MGO(B)、3-DG(C)浓度随加热时间的变化Fig. 7 Changes in the concentrations of GO (A), MGO (B), and 3-DG (C)in the fructose-lysine reaction system with heating time
2.5 茶多酚及其衍生物对戊糖素的抑制作用
戊糖素一般由戊糖和赖氨酸、精氨酸通过Amadori重排后继续氧化交联而产生,是一种具有交联性和荧光特性的糖基化终产物,在其他糖类如果糖、葡萄糖与氨基酸的反应中也可生成[42],可通过荧光强度反映戊糖素的含量[25]。
由图8可知,在反应初期,MA-TP和TP处理组中的戊糖素含量增长速率较慢,随着加热时间的延长,各组别中戊糖素含量明显增加。在整个反应时间内,MA-TP和TP处理组中戊糖素荧光强度显著低于完全糖基化组和阳性对照组(P<0.05),表明MA-TP和TP对戊糖素的生成均具有较好的抑制能力,同时两者对前期抑制效果较好,0.5 h时抑制率分别高达88.43%和84.81%,后期抑制效果可能因样品部分消耗而有所下降。
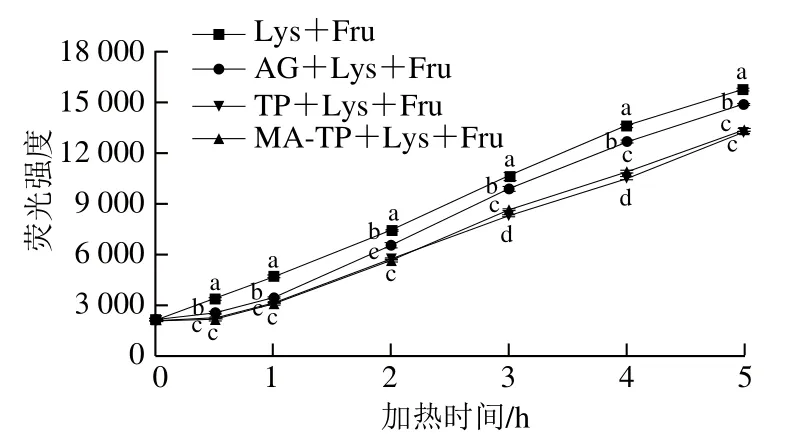
图8 果糖-赖氨酸反应体系中戊糖素含量随加热时间的变化Fig. 8 Changes in the content of pentose in the fructose-lysine reaction system during heating
2.6 茶多酚及其改性衍生物对总体荧光性AGEs的抑制作用
AGEs主要是由蛋白质或氨基酸的游离氨基与还原糖的羰基经过缩合、氧化、重排、交联等一系列反应形成的一类不可逆终产物的总称[43]。由于AGEs多具有自发荧光性,所以可通过测定各反应体系中的荧光强度反映其AGEs水平[44]。
由图9A可知,0~1 h内,Fa对照体系MA-TP+Lys和TP+Lys中荧光强度逐渐提高,随后处于稳定状态,而Fa对照体系AG+Lys和Lys中荧光强度曲线互相重合,并且在加热过程中基本保持不变。MA-TP+Lys组中荧光强度显著高于TP+Lys组(P<0.05),表明经过MA改性后,MA-TP更易与Lys结合。另一方面,从图9B可以看出,在0~2 h内,MA-TP和TP反应体系中荧光性AGEs生成速率缓慢增加,随后迅速增加,这可能是由于反应前期MA-TP和TP与Lys结合,随着反应进行抑制剂逐渐消耗而所导致。同时,比较各组荧光性AGEs的含量,由高到低依次为Lys+Fru>AG+Lys+Fru>TP+Lys+Fru>MA-TP+Lys+Fru(P<0.05),表明经过MA改性,MA-TP对总体荧光性AGEs的生成具有更好的抑制能力,抑制率最高为95.4%(0.5 h时)。这可能是由于MA-TP更易与赖氨酸或Amadori产物结合,占据非酶糖基化的交联位点,导致糖基化后期的交联无法有效进行,从而抑制了荧光性AGEs的形成。

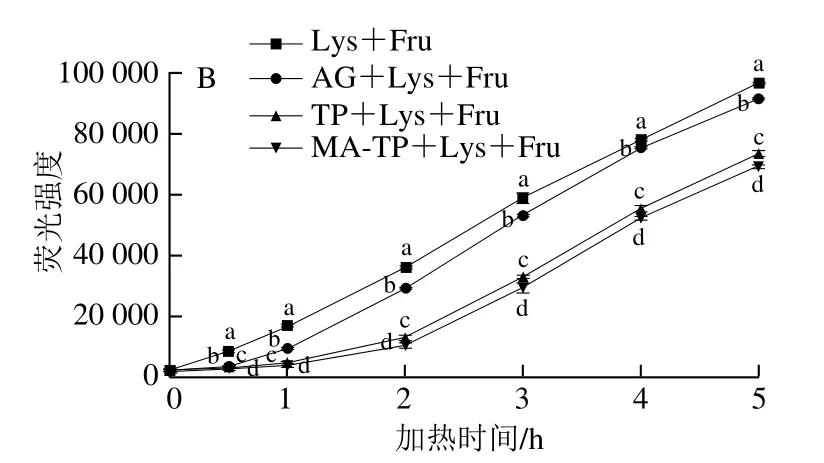
图9 Fa对照体系(A)与样品抑制体系Fs(B)荧光性AGEs含量的变化Fig. 9 Changes in fluorescence intensity of control (A) and inhibition (B)systems during heating
3 结 论
通过MA-TP、TP和AG对Lys-Fru体系中各二羰基化合物(GO、MGO、3-DG)、戊糖素和总体荧光性AGEs抑制能力的实验,证明三者均可抑制糖基化反应。进一步对比发现MA-TP对非酶糖基化抑制作用优于TP和AG,尤其在反应中后期,说明经过MA改性,可以有效提高TP的热稳定性,对抑制高温下的糖基化反应产生积极作用,提高TP的抗糖基化能力。同时,MA-TP易与氨基酸或Amadori产物结合,占据非酶糖基化的交联位点,使糖基化后期的交联无法有效进行,进而抑制了荧光性AGEs的生成。
