富锂锰基三元材料Li1.208Ni0.333Co0.042Mn0.417O2的电子结构和缺陷性质*
2021-12-23黄文军王亚平曹昕睿吴顺情朱梓忠
黄文军 王亚平 曹昕睿 吴顺情 朱梓忠
(厦门大学物理科学与技术学院, 厦门 361005)
(2021 年3 月2 日收到; 2021 年6 月3 日收到修改稿)
1 引 言
锂离子电池自商业化以来, 因具有相对较高的比能量、良好的倍率性能和有效的循环稳定性而得以迅速发展, 并广泛应用于便携式电子产品、电动汽车和航空航天等领域[1-5]. 随着大规模储能应用和技术的迅速发展, 当前的锂离子电池已经不能满足日益增长的对能量密度的要求, 研发更高能量密度的下一代锂离子电池至关重要[6,7]. 电极材料在电池的电化学行为中起着关键的作用, 其中第一个商业化的 LiCoO2正极和碳负极仍然主导着锂离子电池领域[8-10]. 但是, 与已开发的可逆容量超过1000 mA/g 且循环稳定性和低成本的硅/碳复合负极材料相比[11,12], 层状LiCoO2正极实际上只能提供约150 mA/g (其理论比容量可以达到275 mA/g)[9,13]. 此外, 地球上钴源的稀缺性导致LiCoO2材料的高成本, 也限制了它在电动汽车中的应用.
在过去的几十年中, 开发高容量、低成本且具有持久的循环寿命以及优异的充放速率和安全性能的正极材料一直是锂离子电池研究的重要方向.在众多的正极候选材料中, 层状过渡金属氧化物,包括Li(NixCoyMn1–x–y)O2(即NCM 三元材料) 和富锂的Li1+z(Mn1–xMx)1–zO2(M= Ni, Co, Ru, Sn等)[14-16], 尖晶石型的LiM2O4(M= Ni, Mn)[17,18]和橄榄石型的LiMXO4(M= Fe, Mn, Co;X=P, Si)[19-21]正极材料得到了特别的重视. 富锂三元层状氧化物正极材料, 由于其高于300 mA/g 的比容量, 大于1000 W·h/kg 的能量密度以及丰富锰源带来的低成本, 被认为是下一代锂离子电池中最具有应用前景的正极材料之一[22-24]. 本文的研究对象就是一种富锂锰基的三元层状氧化物Li1.208Ni0.333Co0.042Mn0.417O2, 该材料有着高的放电电压、优异的结构和循环稳定性、高的电压/容量保持率[25]. 当它用于包覆其他的富锂三元材料时, 通过表面富Ni 的设计能够增加整个体系的结构稳定性, 抑制其容量减小以及电压衰减[25]. 钴元素不仅是活性材料, 而且可以有效抑制阳离子的混排(Li/Ni) 并稳定材料的结构, 有助于改善材料的深度放电特性并提高倍率性能; 锰元素对材料有支撑作用, 也可以抑制在大量脱锂时阳离子的混排, 但是锰含量的上升会部分降低材料的容量; 此外, 镍元素含量的上升能够提高材料的工作电压, 但由于Ni 在Li 层中较小的扩散势垒更容易在材料表面选择性偏析, 降低材料的结构稳定性[3,26,27]. 通过第一性原理的方法, 本文对Li1.208Ni0.333Co0.042Mn0.417O2材料的结构和电子结构性质进行了仔细的计算,表明了该组分的层状三元材料是一个具有直接带隙的磁性半导体. 通过计算各个过渡金属原子的空位形成能, 探讨了Ni, Co 和Mn 元素在材料中的键合强度, 结果表明Ni 元素最容易从材料中脱出,而Mn 元素最难从材料中脱出. 这些研究能够帮助理解这个具有复杂组分的三元材料的基本性质.
2 计算方法
本文采用的是基于密度泛函理论 (DFT) 的第一性原理计算方法, 通过从头计算的VASP 程序包[28,29]来实现. 该程序包基于平面波展开和投影缀加波 (projector augmented-wave, PAW)[30,31]方法. 计算中, 交换关联泛函使用的是广义梯度近似(GGA)[32]下的PBE (Perdew-Burke-Ernzerhof)[33]表述, 波函数则按平面波基展开, 展开时的平面波截断动能为550 eV. 布里渊区 (BZ) 上的积分替换为倒格空间特殊k点集上的离散求和, 采用了Monkhorst-pack 的5 × 2 × 2k点网格[34]. 采用的超晶胞原胞的晶格常数和原胞内的原子坐标都进行了充分的弛豫, 直到原胞内所有原子上的Hellmann-Feynman 力均小于0.2 eV/nm 为止. 此外, 计算还采用了GGA +U[35]的方法来处理具有局域性特点的Ni, Co 和Mn 的3d 电子的强关联性质, 其中有效相互作用参数Ueff值参考了Jain等[36]报道的数值, 即对Ni, Co 和Mn 原子分别使用了Ueff= 6.0, 3.4 和3.9 eV. 由于过渡金属d 电子的局域性强关联特点, 使用GGA +U方法是必要的. 整个计算是在自旋极化的框架下进行的, 因为材料的电子结构会受到Ni, Co, Mn 原子磁性的明显影响.
3 结果分析与讨论
Li1.208Ni0.333Co0.042Mn0.417O2具有与LiCoO2类似的α-NaFeO2型层状结构, 属于六方晶系, 空间群为. 每个原胞含有24 个单位分子式 (单位分子式可以从LiCoO2来看, 当体系富锂加上三元时即为Li1.208Ni0.333Co0.042Mn0.417O2), 共有96 个原子/原胞, 其中含有29 个Li 原子, 8 个Ni 原子,1 个Co 原子, 10 个Mn 原子以及48 个O 原子, 晶体结构如图1 所示. 可以看出, 所有Li 离子和过渡金属 (TM= Ni, Co, Mn) 离子都处在密堆积氧晶格八面体的中心位置而形成LiO6八面体和TMO6八面体. 作为富锂的层状结构, 如果将Li1.208Ni0.333Co0.042Mn0.417O2重新写为LiTMO2(其中TM=Li0.208Ni0.333Co0.042Mn0.417) 的结构式, 则晶格可看作是紧密堆积的Li-O-TM-O 层的交替排列, 其中的TM层由金属元素和富余的Li 有序镶嵌形成.整个晶体可以看作是由共享边的LiO6八面体层和TMO6八面体层在c方向上的交替堆垛而成,这种结构非常适合于锂离子的嵌入与脱出.

图1 Li1.208Ni0.333Co0.042Mn0.417O2 的晶体结构, 其中LiO6,NiO6, CoO6 和MnO6 八面体分别标识为绿色、灰色、蓝色和紫色Fig. 1. Crystal structure of Li1.208Ni0.333Co0.042Mn0.417O2,where LiO6, NiO6, CoO6 and MnO6 octahedra are marked by green, gray, blue and purple, respectively.
表1 中给出了Li1.208Ni0.333Co0.042Mn0.417O2以及带有过渡金属单空位缺陷材料的晶格常数、原胞体积、体积变化率和空位形成能. 空位的计算时,直接在计算完整晶体 (空位形成前) 的原胞中拿去一个相应的金属原子而形成空位. 所以, 空位计算的超原胞是95 原子/原胞. 从表1 中的结果可以看出, 脱去一个Mn 原子, 体积变小最多; 脱去一个Ni 原子体积变小次之; 而脱去单个Co 原子后, 体积几乎没有变化. 再来看一下单个金属M的空位形成能, 其公式如下:
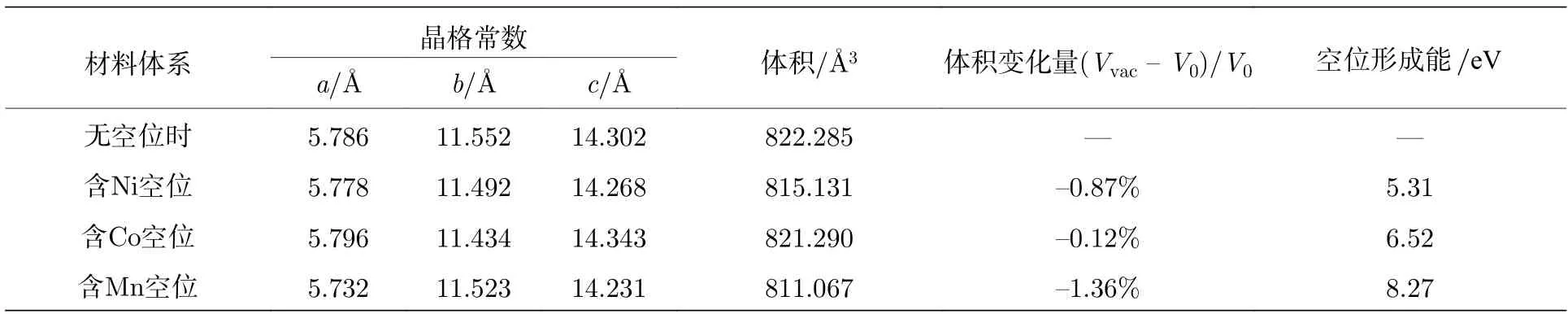
表1 空位形成前后Li1.208Ni0.333Co0.042Mn0.417O2 的晶格常数, 原胞体积和空位形成能的计算值Table 1. Computational lattice constants, unit cell volume and vacancy formation energies of Li1.208Ni0.333Co0.042Mn0.417O2 before and after the defect formations.
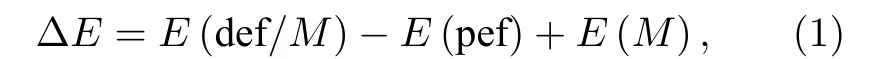
其中E(pef) 表示完整晶体的总结合能,E(def/M)表示脱去单个金属原子后缺陷晶体的总结合能,E(M)为M的体相中单个过渡金属原子的结合能(M= Ni, Co, Mn). 计算结果表明, Mn 空位的形成能最大, 意味着Mn 空位最难形成, 这与体系是锰基材料相一致. Mn 对材料的结构稳定性贡献最大. 此外, Ni 空位的形成能最小, 这意味着Ni 元素最容易脱离材料, 也是三个金属元素中最活跃的.Ni2+离子的半径约为0.69 Å, 这与Li+的半径 0.73 Å相近. 实验结果表明, Ni2+容易跳出TM层而与Li 层的Li+发生交换, 形成阳离子混合[27,37], 这与计算的Ni 空位形成能值较低相一致. 而且前面还提到, Mn 含量的上升有利于提高结构稳定性, 这也与理论计算的Mn 缺陷形成能较大相一致.
Li1.208Ni0.333Co0.042Mn0.417O2材料的电子结构计算是在自旋极化的密度泛函理论框架下进行的,这是因为材料中含有的Ni, Co 和Mn 是3d 过渡金属元素, 这些元素使所构成的材料有强烈的自旋极化倾向. 图2 给出了自旋向上和自旋向下的Li1.208Ni0.333Co0.042Mn0.417O2材料的能带结构图. 从图2可以看出, 自旋向上和自旋向下的能带表现出非常不同的电子行为, 表明该材料是一个磁性半导体.能带结构的价带顶 (VBM) 和导带底 (CBM) 都位于Γ点位置, 所以Li1.208Ni0.333Co0.042Mn0.417O2是一个直接带隙的磁性半导体, 且在Γ点位置的带隙为0.47 eV.
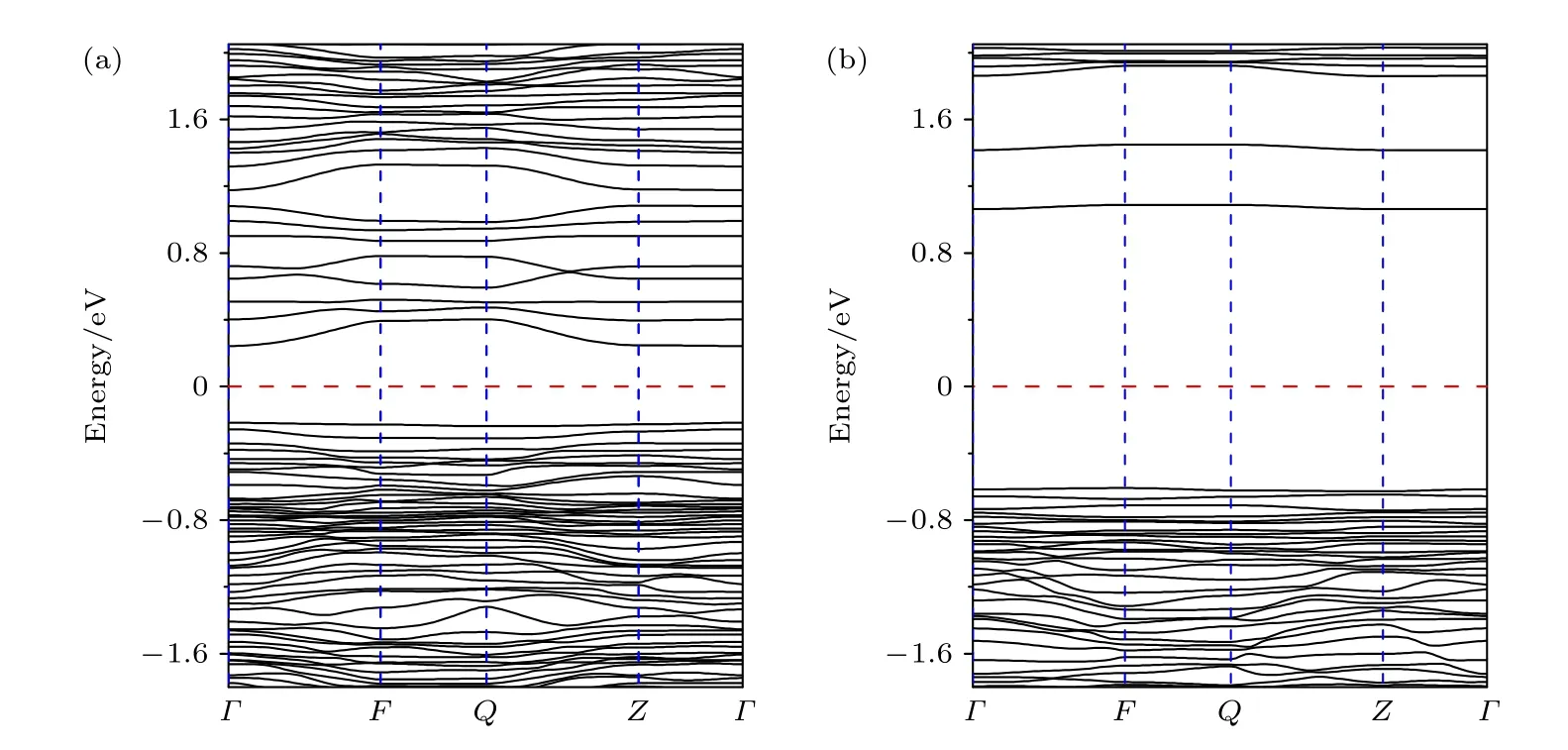
图2 Li1.208Ni0.333Co0.042Mn0.417O2 的能带结构 (a) 自旋向上的能带; (b) 自旋向下的能带. 红色虚线表示费米能级Fig. 2. Band structures of Li1.208Ni0.333Co0.042Mn0.417O2: (a) Spin-up bands; (b) spin-down bands. Fermi level represented by the red dotted line.
这里重点分析价带顶和导带底的电子波函数的电子轨道贡献 (即来自各原子各电子轨道的贡献情况), 具体表达式为
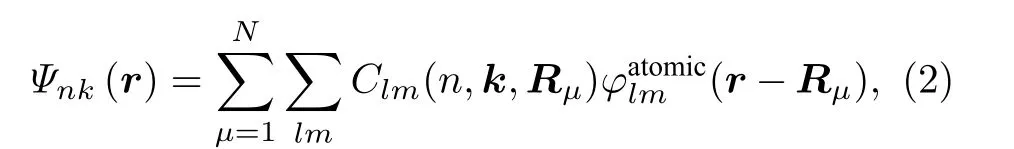
式中, 左边是 (n,k) 态的电子波函数, 右边是该波函数对各原子的各电子轨道r −Rµ) 的展开, 展开系数Clm(n,k,Rµ) 表达了某个电子轨道对某波函数的贡献程度. 表2 列出了价带顶和导带底的电子轨道的贡献. 由表中结果可见, 价带顶的电子态主要是O 的2px, 2py, 2pz轨道与Ni 的3dxy, 3dyz, 3dxz原子轨道之间的杂化, Co 和Mn 原子的电子轨道基本上没有参与; 导带底电子态的原子轨道贡献除了有价带顶的特征外, 还有部分的Ni- 3dx2−y2和Mn- 3dx2−y2, Mn- 3dyz轨道参与杂化.由过渡金属中贡献最多的是Ni 的3d 轨道电子可见, Ni 在Li1.208Ni0.333Co0.042Mn0.417O2材料的过渡金属中电子性质最为活泼. 这与各过渡金属原子在三元材料中不同价态的能级排列特点[38]是一致的(即 Ni2+, Co3+, Mn4+费米面下的能级顺序分别是越来越远离费米面的).
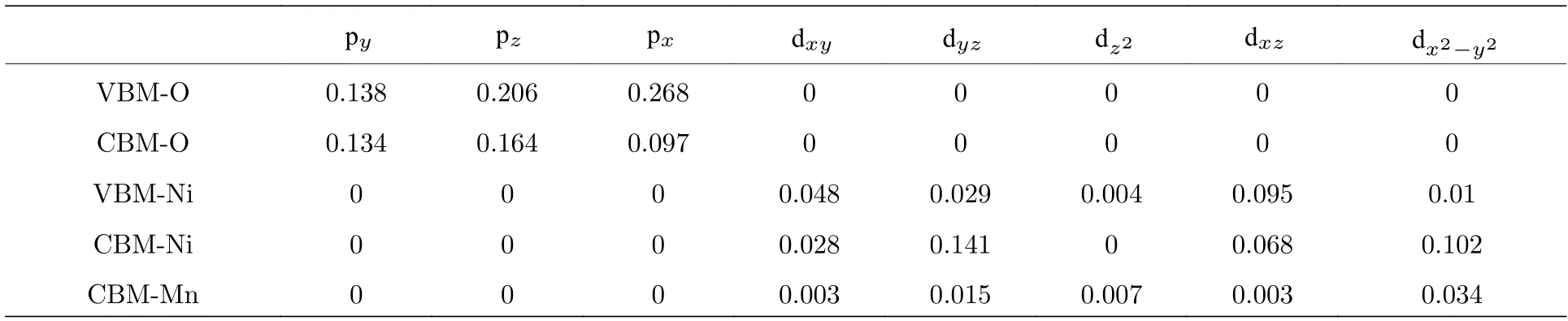
表2 各原子的电子轨道对价带顶和导带底上电子态的贡献Table 2. Contribution of electronic orbital of various types of atoms for the electronic states at VBM and CBM.
计算得到的Li1.208Ni0.333Co0.042Mn0.417O2材料的总电子态密度 (TDOS) 和各原子分态密度 (PDOS)如图3(a)所示. 从图3(a)可以清楚地看到, 该材料是一个半导体, 而且是一个自旋极化的磁性半导体. 在图3(b)—图3(d)中, 分别给出了Li1.208Ni0.333Co0.042Mn0.417O2中空位周围的6 个O 原子的2p电子分态密度和在过渡金属空位 (即Ni, Co 和Mn 空位) 形成之前和形成之后的对比. 可以看到,当金属空位形成后, 费米面附近的O-2p 占据态(DOS 峰) 明显增加. 这是因为, 金属空位的形成打断了金属与周围O 原子的成键, 导致周围这些氧原子上的电子更加自由, 能量上就更加接近于费米面. 带有Co, Ni 空位的体系的态密度特征类似, 即费米面附近有了很高的态密度值 (较自由的电子较多). 而Mn 空位体系的DOS 稍有不同, 其高态密度的峰离费米面稍远 (电子被束缚的较紧).
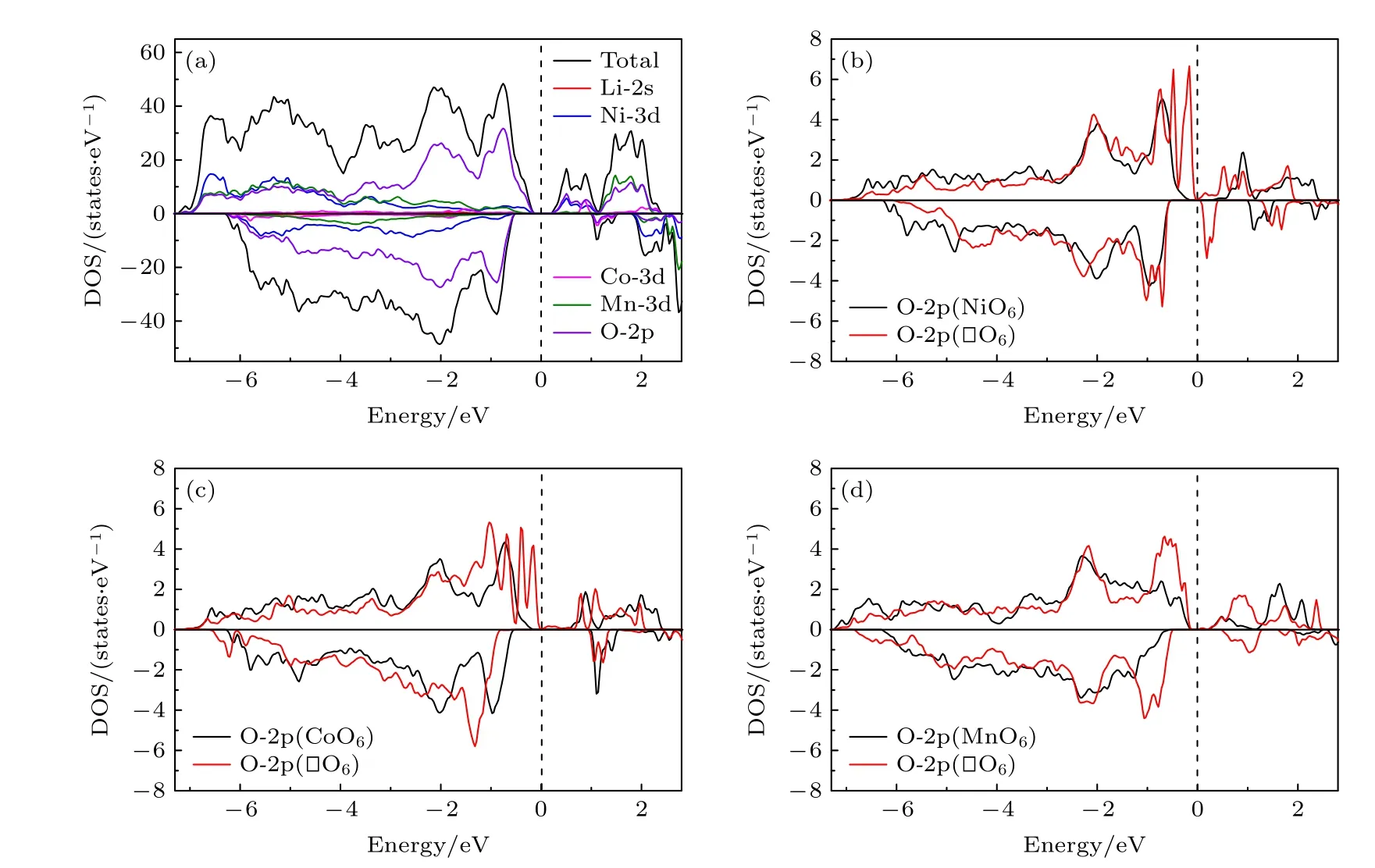
图3 (a) Li1.208Ni0.333Co0.042Mn0.417O2 材料的总态密度和各原子分态密度; (b)—(d) 金属空位形成前后材料中空位周围的6 个氧原子的2p 电子态密度和, 分别用黑色和红色实线表示Fig. 3. (a) Total and atomic-decomposed partial density of states for Li1.208Ni0.333Co0.042Mn0.417O2; (b)–(d) sum of the density of states of 2p electronic states of the six oxygen atoms around the M-vacancy before and after the formation of M vacancy, respectively.Black and red solid lines represent DOSs before and after the vacancy formation, respectively.


图4 材料中三种过渡金属差分电荷密度对比 (a)—(c) 空位形成前; (d)—(f) 空位形成后. 所画的平面是金属附近4 个O 所在的平面. 图中实线和虚线分别表示电荷聚集 (Δρ>0) 与电荷移出(Δρ<0)Fig. 4. Contour plots of the charge density differences: (a)–(c) Before the M-vacancy formation; (d)–(f) after the M-vacancy formation. The solid lines and the dot lines represent the charge accumulation (Δρ > 0) and the charge depletion (Δρ < 0), respectively.
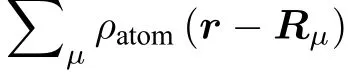
4 结 论
本文基于密度泛函理论的第一性原理方法, 计算了R¯3m相的富锂锰基三元材料Li1.208Ni0.333Co0.042Mn0.417O2及其含过渡金属原子缺陷体系的晶体结构、电子结构和缺陷性质. 计算的能带结构表明, 该材料为直接带隙的磁性半导体, 在Γ点的直接带隙为0.47 eV. 价带顶上的电子轨道贡献主要来自O-2p 和Ni-3d 的轨道电子, 而Co 和Mn原子的3d 电子基本上没有贡献; 导带底上的电子轨道贡献也主要来自O-2p 和Ni-3d 的轨道电子,但此时 Mn 的 3dyz和 3dx2−y2的轨道电子也有部分参与. 计算结果还表明, 锰空位形成时的体积变化率最大且空位的形成能也最大, 意味着Mn 空位最难形成, 而且对材料的结构稳定性贡献最大.Ni 空位的形成能最小, 这意味着Ni 元素最容易脱离材料的. 差分电荷密度的计算显示, 金属原子与O 原子之间的成键方式主要是共价键和离子键的混合. 金属空位的产生仅强烈影响空位附近O 原子周围的电荷密度分布, 这体现了金属空位影响的局域性特征. 本研究可以为理解富锂锰基三元正极材料的组分、结构、电子结构和缺陷性质提供参考.
