基于Bagging集成学习的蛋白质折叠识别
2021-12-17杨欣华顾海明
杨欣华,顾海明
(青岛科技大学数理学院,山东青岛266061)
蛋白质在生物体的生命活动中起着非常重要的作用,而蛋白质的功能取决于蛋白质折叠以及与其他蛋白质的相互作用[1]。蛋白质折叠识别是从蛋白质的氨基酸序列中得到蛋白质的三级结构而不依赖于蛋白质序列的相似性[2]。在人体中蛋白质会发生折叠错误而引起很多的疾病。而深入了解蛋白质折叠对于这些疾病的致病机制,阐明蛋白质错误折叠的各种因素将有助于医学研究和医学药物的研发。但目前的试验方法的代价高,而计算方法具有比实验室方法更便宜更快的优点,现在普遍用于蛋白质折叠识别[3]。目前对蛋白质折叠的研究是根据蛋白质主要结构信息将蛋白质进行分类,由此得到已知的蛋白质折叠类型[4]。基于分类的方法就是基于各种蛋白质结构分类数据库中的数据运用机器学习方法对蛋白质进行结构分类。
目前得到所需要的蛋白质折叠信息主要是是通过机器学习,从蛋白质序列出发对蛋白质折叠模式的分类。常用的数据库有蛋白质结构分类数据库SCOP(structural classification of protein,SCOP)数据库[5]。其中SCOP 数据库包括蛋白质结构类、折叠类型、超家族、家族等不同层次[6],本研究所使用的数据集同样是从SCOP 数据库中选取的。蛋白质折叠识别作为多类分类任务,在该领域中已经有很多基于机器学习方法构建的模型。这些方法中的大多数包含两个阶段:1)特征提取;2)分类算法[4]。
对于特征提取方法也有许多,例如位置特异性得分矩阵 (position specific scoring matrix,PSSM)[7]、分组重量编码(encoding based on grouped weight,EBGW)[8-10]、伪氨基酸组成(pseudo amino acid composition,Pse AAC)[11-13]来提取特征信息;例如预测蛋白质序列的二级结构有助于提高分类精度[14]。研究表明,各种特征提取方法的信息是互补的。对于分类的方法,也有许多分类器用于蛋白质结构分类预测。2017年XIA 等[6]提出基于模板分配和支持向量机(support vector machine,SVM)相结合的集成方法[15]。2018 年SUDHA等[16]提出的一种新的增强型人工神经网络(artificial neural networks,ANN)模型下得到了较高的预测的结果。2019 年YAN 等[4]提出MV-fold 与两种基于模板的方法H Hblits(HMM-HMM-based lightning-fast iterative sequence search,HHblits)方法和基于隐马尔可夫模型(hidden Markov model,HMM)的HMMER 方法相结合的方法。虽然目前取得了一定的研究成果,但是仍有很大的研究空间。如何选择计算效率高花费少,性能好的预测方法是研究的重点。
集成分类器通过多个基分类器组合在一起,会获得比单一学习器优越的泛化性能。目前,已有许多的集成分类器用于分类算法,并且可以得到较好的预测结果。本研究提出一种用于蛋白质折叠识别的BAG-fold 模型。在BAG-fold模型中使用4 种不同的特征提取方法提取蛋白质序列的特征信息,并将4种蛋白质特征信息进行融合。并使用LFDA方法处理融合后的特征空间,选取更有效的特征信息。将降维后得到的最优特征子集输入到集成分类器中,通过集成分类器来进一步提高分类准确性。根据10折交叉验证得到的预测结果进行蛋白质折叠识别。实验结果表明,本研究的方法可以达到较好的预测性能。
1 材料与方法
1.1 数据集
在本研究中使用两个数据集来评估构建的模型在折叠识别中的性能。两个数据集包括DD 数据集[2],RDD 数据集[17]。DD 数据集获自蛋白质结构分类SCOP 数据库,DD 数据集包含695个蛋白质序列,具有27个折叠类别,并且数据集的序列同源性小于35%。DD 数据集中的4个主要类是α,β,α+β和α/β。RDD 数据集是DD 数据集的修订版。包含了691个蛋白质序列,具有27个折叠类别。详细信息如表1所示。

表1 DD数据集与RDD数据集的折叠类别Table 1 Folding categories of DD dataset and RDD dataset
1.2 伪位置特异性得分矩阵
位置特异性得分矩阵(position specific scoring matrix,PSSM),利用迭代PSI-BLAST[7]搜索方法提取蛋白质序列信息,得到它对应的PSSM 是L×20(其中L为蛋白质序列的长度)的矩阵
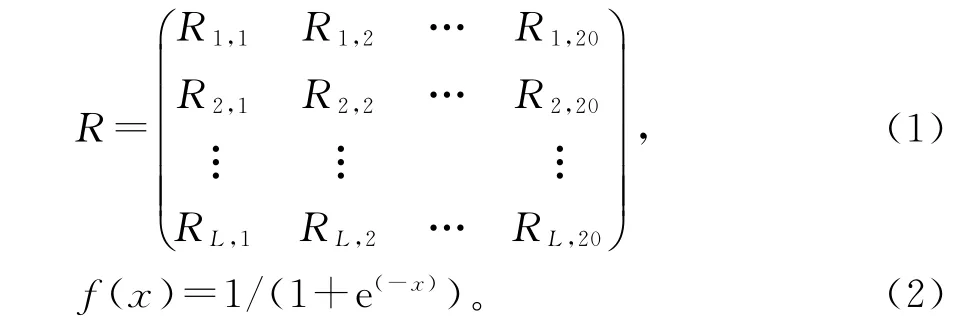
利用公式(2)把PSSM 矩阵中的元素转化到0~1之间。当数据集中含有长度不等的蛋白质序列时,则蛋白质序列的PSSM 矩阵应转化为维数相同的向量。PsePSSM 特征提取[18-19]结果:
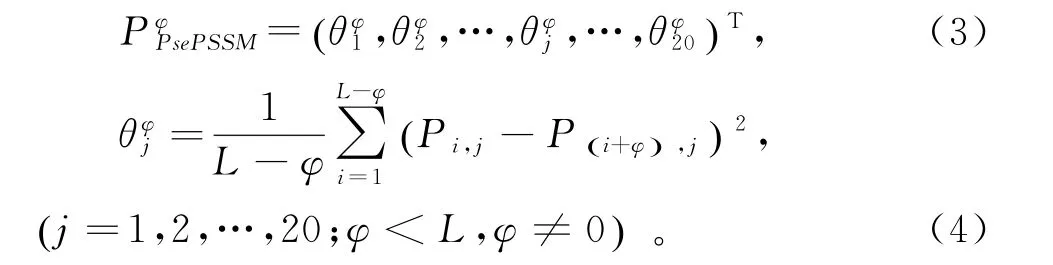
其中,表示氨基酸j的φ阶相关因子,用PsePSSM 表示蛋白质序列为
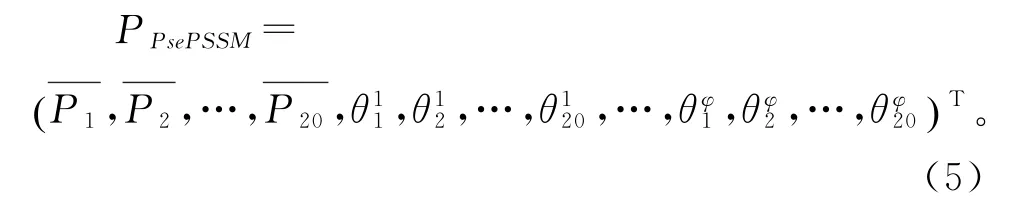
由此一条蛋白质序列PsePSSM 生成一个20+20×φ维的特征向量。
1.3 去趋势互相关分析法
DCCA coefficient是为了量化两个非平稳时间序列之间的互相关水平。每个蛋白质序列的PSSM矩阵的大小是L×20,将PSSM 矩阵中的20列作为20个非平稳时间序列进行计算[20]。
在对PSSM 矩阵标准化后,对于任意PSSM的两个不同列{pi}和{qi},i=1,2,…,L,L表示蛋白质序列的长度。首先利用公式(6)计算新的时间序列P和Q。
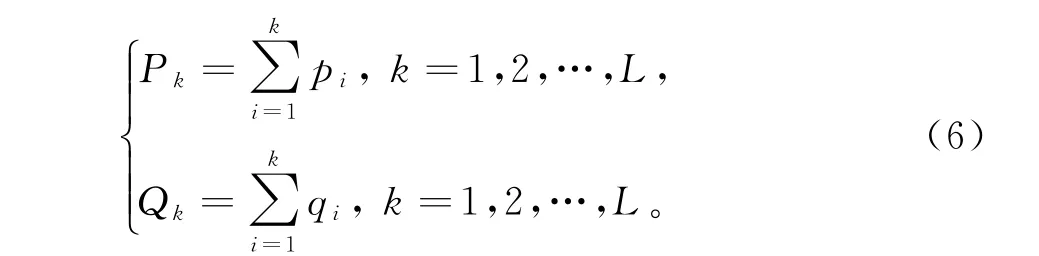
然后把Pk和Qk分成L-T段可重叠的部分,每段含有T+1个数据,并对每段进行最小二乘线性拟合,则拟合值为。利用公式计算每一段的协方差
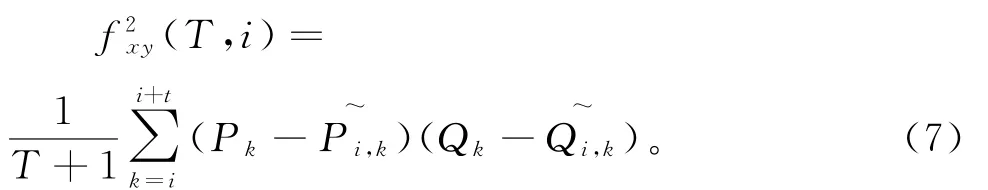
接下来,利用公式(8)计算L-T段的协方差
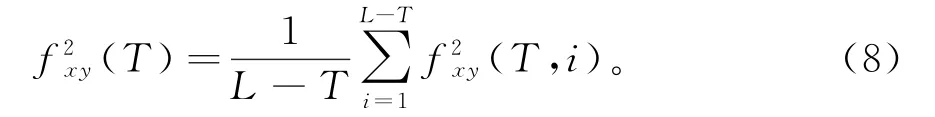
最后,利用公式(9)计算两个不同时间序列{pi}和{qi}的DCCA coefficient。取值范围为-1≤ρDCCA≤1,因此,1表示完全的互相关,0表示没有互相关,-1表示完全的反互相关。通过DCCA coefficient算法将一条蛋白质序列生成190维的特征向量。
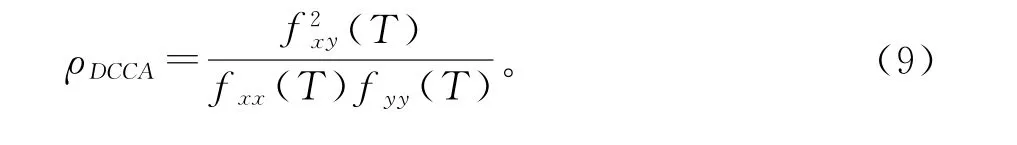
1.4 二级结构
二级结构是通过骨架上氨基之间形成的氢键定义的。SS的统计表示基于3 种状态构建:α-螺旋(α-helix,H),β-链(β-strand,E)和无规卷曲(random coil,C)。在网址https://sparks-lab.org/server/spider3/上进行SPIDER3 预测可以得到[21],可以定义此功能:
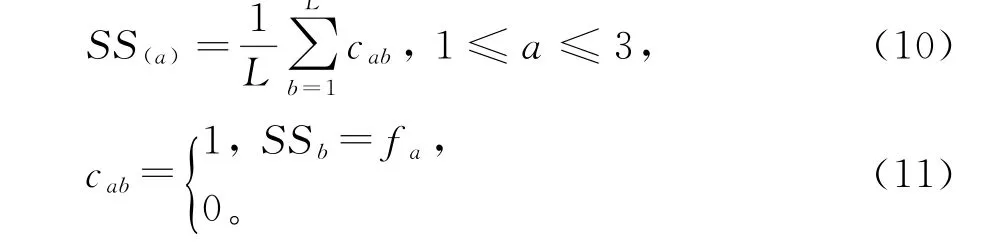
其中,L是蛋白质的长度,fa是H,E和C。
1.5 分组重量编码
ZHANG 等[22]提出了EBGW 方法对蛋白质序列进行特征提取。由氨基酸对应的物理化学性质分成四类:碱性氨基酸U1={H,K,R}、酸性氨基酸U2={D,E}、中性和极性氨基酸U3={Q,N,S,T,Y,C}和中性和疏水性氨基酸U4={G,A,V,L,I,M,P,F,W}。将4 种划分方式两两合并可以得到{U1,U2}和{U3,U4}、{U1,U3}和{U2,U4}、{U1,U4}和{U2,U3}。假设一条蛋白质序列P=p1p2p3…pN,经过划分后:
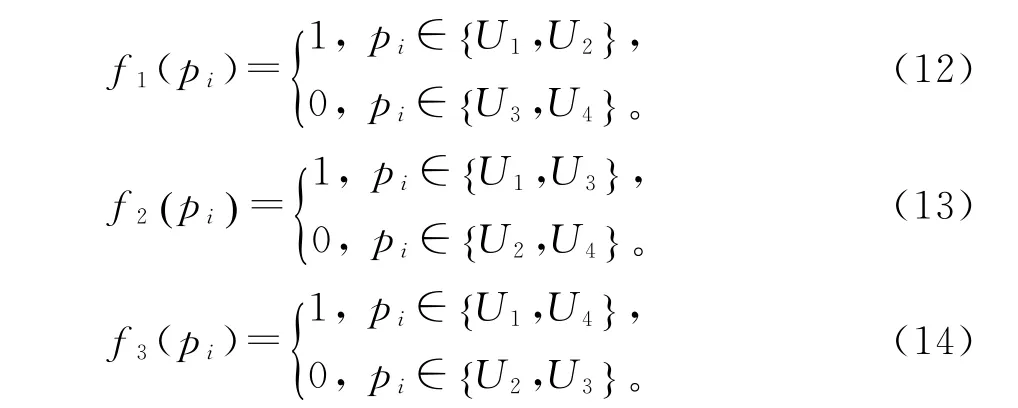
利用公式(12)、公式(13)、公式(14)将K序列P变成3 条长度为N的二进制序列G1(P)、G2(P)、G3(P)。把这些序列划分成若干个长度依次递增的子序列,设定一个固定参数M,子序列可以表示为kn/M」,k=1,2,…,M。其中·」表示取整运算符,计算每一条子序列中1出现的频率,一条Gi(P)可以转化成特征向量。综上,对于一条长度为N的蛋白质序列,可以得到3M维向量。
1.6 局部Fisher判别分析
局部Fisher 判别分析(local Fisher discriminant analysis,LFDA)[23]是一种有监督的降维方法。令蛋白质数据矩阵为X=x1,x2,…,xn
[],其中,n是蛋白质的样本个数,d是蛋白质序列特征提取的维数。由公式(15)计算局部类内散射矩阵H(w)和局部类间散射矩阵H(b):
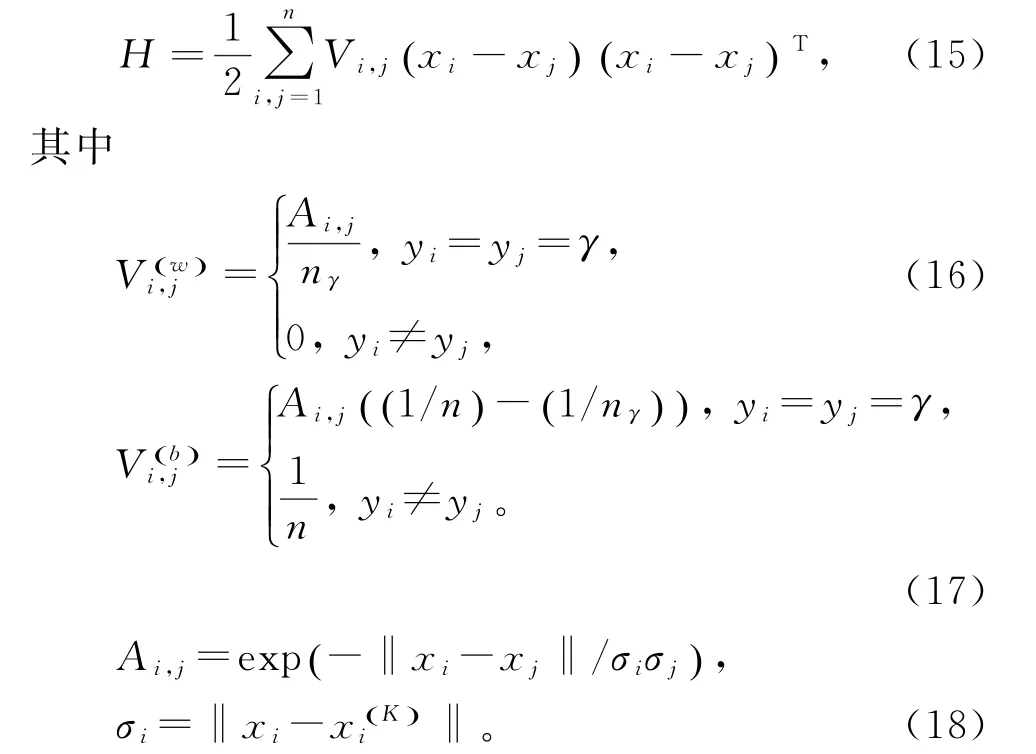
其中,A是一个亲和度矩阵,Ai,j∈A是xi和xj的亲和度。亲和度矩阵(18)表示围绕xi的数据样本的局部缩放,其中x(K)i是xi的第K个最近邻。
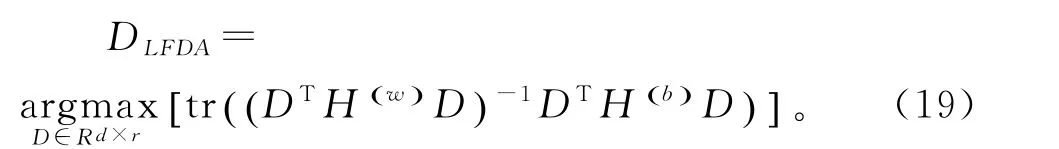
降维后的矩阵变为

1.7 集成学习
在机器学习的有监督学习算法中,预测效果好并且稳定的模型是不容易得到的。而集成学习则是结合多个弱监督模型来得到效果比单个弱监督模型更好的强监督模型,集成学习是用多个弱分类器来纠正某一个弱分类器错误的预测。集成分类器系统的框架是通过将众多基本分类器组合在一起而建立的,而建立起的框架可以减少因单个训练集的特殊性而引起的方差。假设基分类器计算相应的复杂度为Ο(m),则Bagging的计算复杂度T(Ο(m)+Ο(s)),由于采样与投票/平均的复杂度Ο(s)很小,且T是较小的一个常数,所以训练一个Bagging集成与直接使用基学习算法训练分类器的复杂度同阶,这更加能说明Bagging是一个高效的集成学习算法。本研究中用的是Bagging算法,是由决策树集成的同种类型基分类器的集成。算法过程如下[24]:
1)从原始样本集中抽取训练集。D=(x1,y1),(x2,y2),…,(xn,yn){}每次使用Bootstraping方法从原始样本集中抽取n个训练样本。共进行k轮抽取,得到k个训练集。其中k个训练集之间是相互独立的。
2)每次用一个训练集训练出一个模型,k个训练集共训练出k个模型。
3)将2)得到的k个模型采用投票的方式得到分类结果(21)。
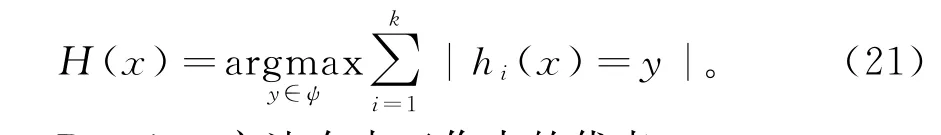
Bagging方法在本工作中的优点:
1)每个模型独立构建。各个基分类器之间相互独立。
2)能够减少方差。整体模型的方差小于等于基模型的方差,随着基模型数的增多,整体模型的方差减少,从而防止过拟合的能力增强,模型的准确度得到提高。
3)适用于高方差低偏差模型。对于Bagging算法来说,算法会并行地训练很多不同的分类器,然后采用多数投票原则/平均值原则。所以在同一样本上训练出来的模型方差小。
4)Bagging方法是相对于单个模型改进的一种方法,无需调整底层的基本算法。
5)提供了一种减少过度拟合的方法。
1.8 模型评估
在本研究中采用10折交叉验证来检验预测模型的性能。本工作采用准确率(accuracy,Acc)、敏感性(sensitivity,Sen)、特异性(specificity,Spe)、Matthew 相关系数(MCC)和精确率(precision,Pre)来评价预测模型的结果。指标定义如下:
准确率是正确预测的蛋白质的数量与所研究的蛋白质的总数的比率:
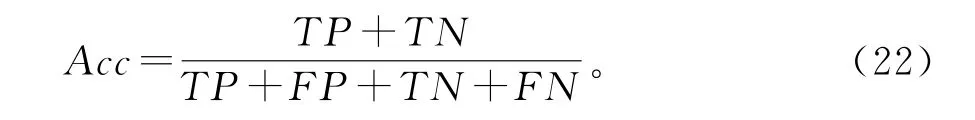
特异性是正确剔除样品与剔除测试样品总数的比率:

灵敏度是对每种类别的测试数据集中正确分类的样本与整个样本的比率:

精确率是精度测量正确分类的样本与整个阳性预测样本的比率:

整体上衡量模型性能的Matthew 相关系数:
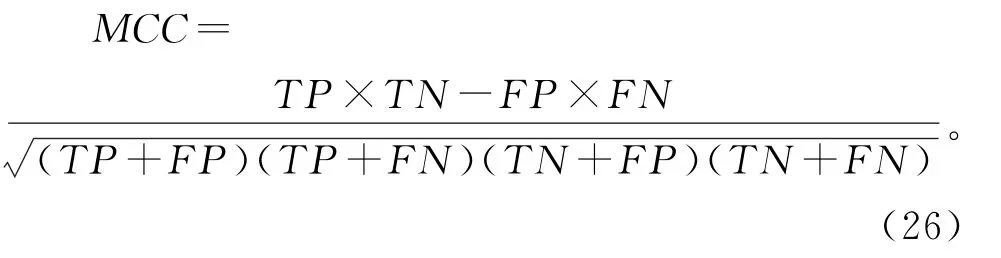
其中TP为真阳性样本,FN为假阴性样本,TN为真阴性,FP为假阳性。本工作使用由精确度、特异性、灵敏度和Matthew 相关系数数组成的3个标准来证明本工作的方法在DD 和RDD 数据集上的性能。
为了方便起见,提出的蛋白质折叠的预测方法BAG-fold的流程如图1所示。
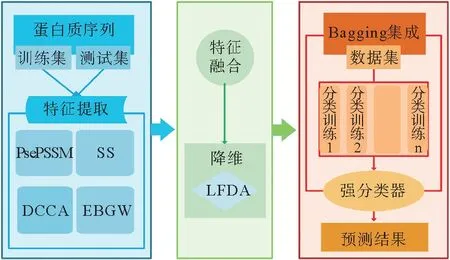
图1 基于BAG-fold方法的蛋白质折叠预测流程图Fig.1 Flow chart of protein folding prediction based on BAG-fold method
基于BAG-fold方法的蛋白质折叠识别的步骤可以描述如下。
1)从SCOP中获取数据集,输入数据集中的蛋白质序列和相对的类别标签。
2)特征提取。将蛋白质序列通过编码将字符信号转化为数值信号。利用DCCA、PsePSSM、SS、EBGW 对蛋白质信息进行特征提取。然后将4 种特征提取的特征进行融合,数据集中每条蛋白质序列由590维的数值向量表示。
3)降维。使用LFDA 算法去除特征融合后产生的冗余和噪声,筛选最优的特征子集,从而为输入分类器提供良好的特征信息。
4)根据步骤2)和3),将所选取的最优特征子集及其所对应的标签,输入到Bagging集成分类器中结合10折交叉验证进行蛋白质折叠识别。
5)模型性能评估。将Acc、Sen、Spe、Pre和MCC作为评价指标,检验模型的预测性能。
2 结果与讨论
2.1 参数选择
参数的选取是从蛋白质序列中提取有效的特征信息的关键的一步,参数的选取对模型的构建起着非常重要的作用。为了得到更好的特征信息,需要不断地对参数进行调整。在本工作中,通过10折交叉验证调节参数得到一组最佳的模型参数。本工作把训练集DD 作为研究对象,对蛋白质序列进行特征提取。本研究方法中有3种方法的参数选择分别为DCCA、PsePSSM 和EBGW。
2.1.1 DCCA 算法的参数值T的选取
采用DCCA coefficient特征提取中T取值不同对模型的预测性能所产生的影响效果也不同。使用参数T确定DCCA coefficient的每一段重合部分的长度。因为两个数据集中蛋白质序列中最短长度为13,因此为了寻找最优参数将DCCA coefficient中T取值依次设置为1、2、3、4、5、6、7、8、9、10、11和12,在集成分类器中进行预测,采用10折交叉验证对结果进行检验。选择不同参数所得到的结果如表2所示。
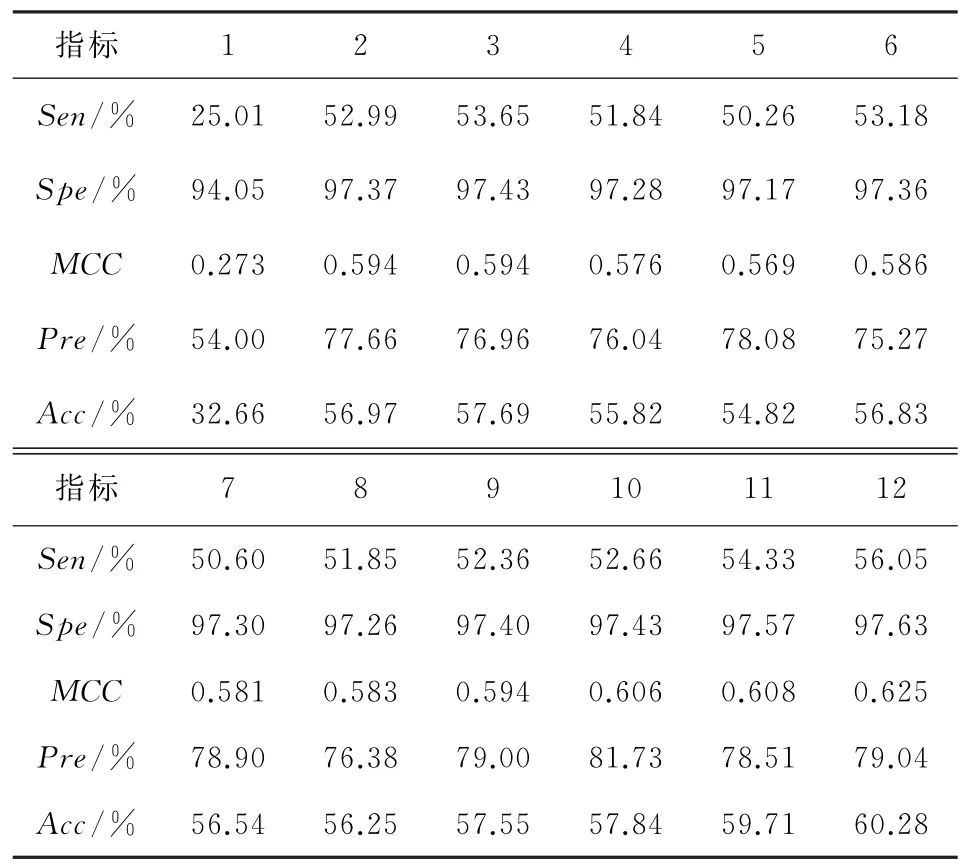
表2 DD数据集对于不同的T 值的结果Table 2 DD data set results for different T values
由表2可知,随着T值的变化,预测的准确率也随之变化。选择值达到最大时所对应的T值作为DCCA 编码中的最佳参数,此时模型的性能最好。由表2可知当T=12时得到的精度是最高的,为60.28%,比其它参数高出0.57%~27.62%。因此本工作DD 数据集选用DCCA的参数值为12。此时特征空间所对应的维数为190维。因此测试集RDD 数据集的参数值同为12。
2.1.2 PsePSSM 算法的参数值φ的选取
PsePSSM 编码的参数φ取值不同对模型的预测性能有一定的影响。两个数据集中蛋白质序列中最短长度为13,因此为了寻找最优参数将PsePSSM的参数φ取值依次设置为1、2、3、4、5、6、7、8、9、10、11和12。在集成分类器中进行预测,采用10折交叉验证对结果进行检验。选择不同参数所得到的结果如表3所示。
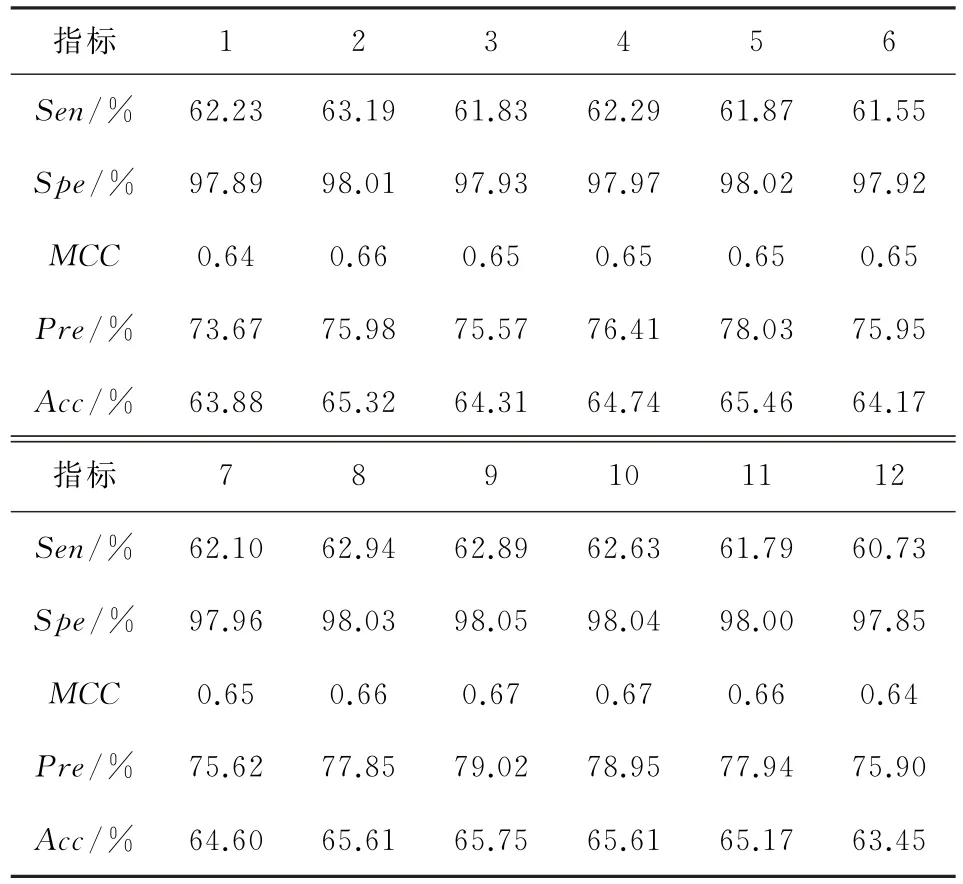
表3 DD数据集对于不同的φ 值的结果Table 3 DD data set results for differentφvalues
由表3可以看出,评价指标随着φ取值的变化而发生变化。当训练数据集中φ取值为9时,数值达到最大,代表此时PsePSSM 算法对模型性能的影响最好,因此选择φ取值为9作为PsePSSM 编码中的最佳参数,φ=9 时PsePSSM的Acc值为65.75%。比其它参数高出0.14%~2.3%。因此本工作训练集DD 选用PsePSSM的参数值为9。因此测试集RDD 数据集的参数值同为9。此时,特征空间所对应的维数为200维。
2.1.3 EBGW 算法的参数值M的选取
EBGW 编码中参数M取值不同对模型的预测性能有不同的影响。两个数据集中蛋白质序列中最短长度为13。因此为了寻找最优参数将EBGW 中参数M的取值依次设置为1、2、3、4、5、6、7、8、9、10、11和12。在集成分类器中进行预测,采用10折交叉验证对结果进行检验。选择不同参数所得到的结果如表4所示。
由表4可以知,评价指标随着M取值的变化而发生变化。当M取值为9时数值达到最大,代表此时EBGW 算法对模型性能的影响最好,因此选择EBGW的最佳参数为9。当M值取值为9时,EBGW的Acc值为36.83%。比其它参数高出0.72%~8.35%。因此训练集DD 选用EBGW的参数值为9。因此测试集RDD 数据集的参数值同为9。此时,特征空间所对应的维数为27维。
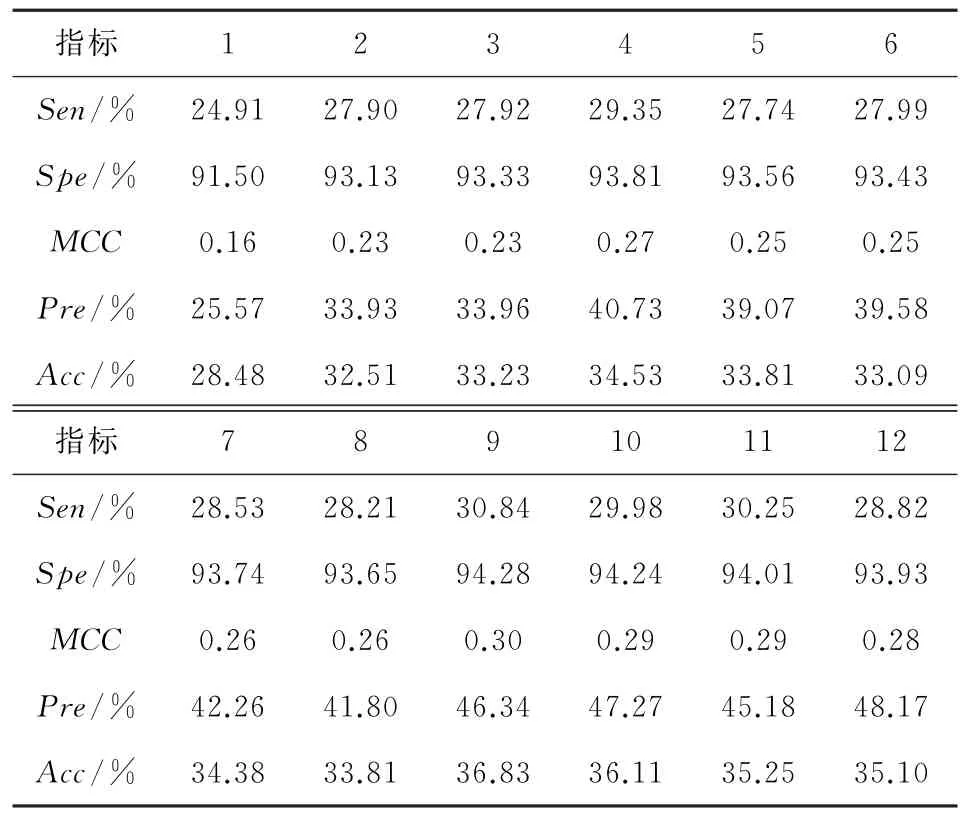
表4 DD数据集对于不同的M 值的结果Table 4 DD data set results for different M values
2.2 特征提取算法对预测结果的影响
使用特征提取算法提取蛋白质序列的有效信息,是构建蛋白折叠识别预测模型的一个重要步骤。本工作共选择了4 种特征提取算法,DCCA、PsePSSM、SS和EBGW。为了更好的研究模型的性能,本文做了如下对比,包括4种单独特征提取方式和特征融合后的方式ALL(PsePSSM、DCCA、SS和EBGW)。将不同的特征提取方式数据集上获得的特征空间分别输入到Bagging集成分类器中进行预测,得到了DD数据集的Acc、Sen、Spe、Pre和MCC值。不同特征提取方式得到的结果如表5和表6所示。
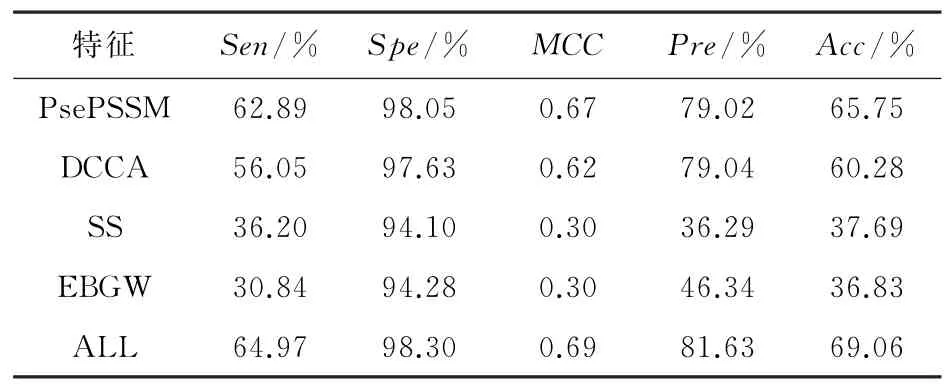
表5 DD数据集不同特征的预测结果Table 5 Prediction results of different features of the DD dataset
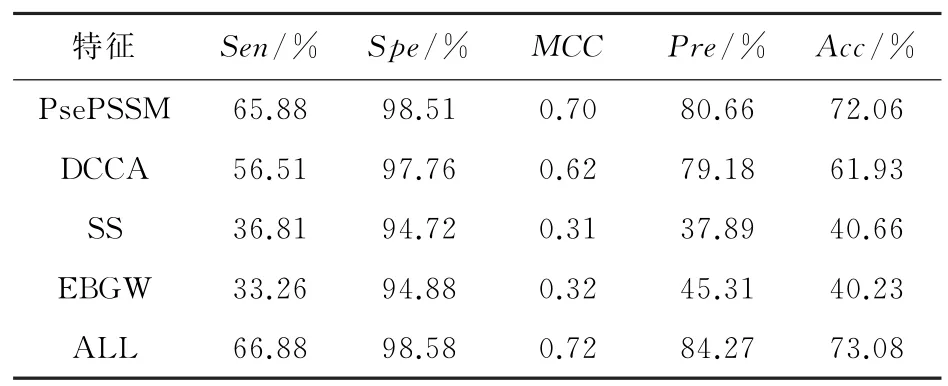
表6 RDD数据集不同特征的预测结果Table 6 Prediction results of different characteristics of the RDD data set
由表5可知,DD 数据集用PsePSSM、DCCA、SS和EBGW 特征提取方式得到Acc值分别为65.75%、60.28%、37.69%和36.83%。而DD 数据集的ALL特征提取方法融合后得到的Acc值为69.06%。由表6 可知,RDD 数据集用PsePSSM、DCCA、SS和EBGW 特征提取方式得到Acc值分别 为72.06%、61.93%、40.66% 和40.23%。而RDD 数据集的ALL 特征提取方法融合后得到的Acc值为73.08%。很明显得到特征提取融合后的结果比单个的结果的Acc值要更高,所以融合后特征与单一特征提取方法相比具有良好的预测性能。
2.3 降维方法对预测结果的影响
通过PsePSSM、DCCA、SS和EBGW 4种特征提取方式对DD 数据集进行特征提取,从序列不同角度提取特征信息,融合后得到419 维特征向量。由于融合后的特征向量维数大并且有冗余所以对数据的准确率有影响,因此,消除冗余信息并且保留原有数据的有效信息显得至关重要。选取不同的特征选择方法对蛋白质折叠识别的准确性有一定的影响。对得到的数据分别通过5种降维方法在分别在Bagging集成分类器中进行测试。5种降维方法分别为最小冗余最大相关(max-relevance and min-redundancy,m RMR)[25]、最小二乘特征选择(leastsquares feature selection,LSFS)[26]、多级特征选择(maximum-likelihood feature selection,MLFS)[27]、遗传算法(genetic algorithm,GA)[28]和局部Fisher判别分析(linear fisher discriminant analysis,LFDA)。降维后的特征向量维数为200。
使用不同的特征选择方法,并根据10折交叉验证在DD 数据集上预测得到的结果如表7和图2。
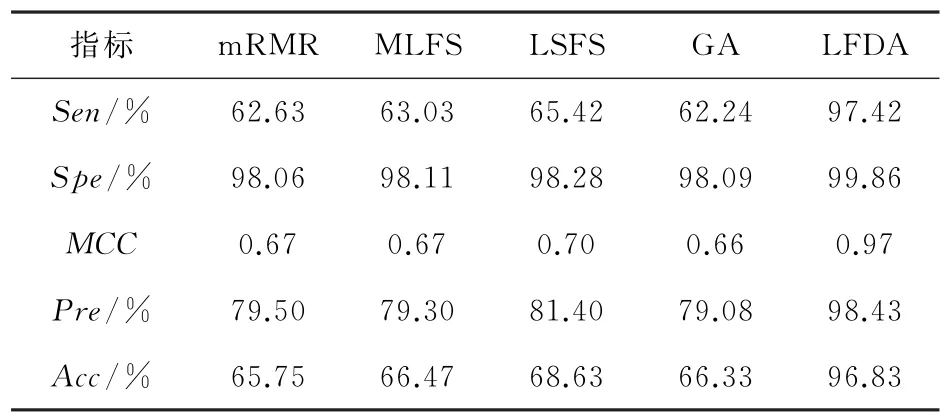
表7 不同降维方法的对比Table 7 Comparison of different dimensionality reduction methods
由表7可知,LFDA 这一降维方法的Acc精度得到了较高的结果。其中LFDA的Acc值比mRMR方法的高出31.08%,比MLFS高出30.36%,比LSFS高出28.2%,比GA高出30.5%。而且使用LFDA 降维时Sen、Spe、Pre和MCC都达到了很高的数值,分别为97.42%、99.86%、98.43%和97%。由图2可以明显的看出使用LFDA 降维方法比使用其它方法的预测结果高。综上所述,使用LFDA 降维方法取得Acc的精度要高于其它的5种降维方法,再次验证了LFDA降维的优异性能。因此本研究选择LFDA 这一降维方法。

图2 LFDA降维方法与不同降维方法进行比较Fig.2 Comparison of LFDA dimensionality reduction method with different dimensionality reduction methods
2.4 与其它分类器进行比较
目前,已经有很多的人对蛋白质折叠识别进行了各式的研究。为了证明BAG-fold 模型的有效性,将预测模型与其它使用相同数据集的预测模型进行比较分析。将模型的性能与其它机器学习方法进行了比较,以展示本研究方法的有效性。与支持向量机(support vector machine,SVM)[29]、K 近 邻(K-nearest neighbor,KNN)[30]、决策树(decision tree,DT)[31]和朴素贝叶斯(naïve Bayes,NB)[32]5种机器学习方法进行了比较。其中SVM 分类器的核函数为sigmoid,其它参数为默认参数;K 近邻分类器的分类中,最近邻的个数为7,计算数据间距离为曼哈顿距离,其它参数为默认参数。DT 和NB分类器都为默认参数。并采用10折交叉验证方法对预测结果进行检验结果,如表8所示。
由表8可知,使用Bagging方法明显比其它的分类器得到的结果好。运用Bagging集成算法作为分类器得到DD 数据集的Acc值最高,达到了96.83%,比SVM 分类器高3.31%,比KNN 分类器高2.59%,比DT 分类器高14.68%,比NB分类器高7.34%。显然,使用Bagging集成算法作为分类器得到的预测结果是最高的。因此基于Bagging集成分类器的BAG-fold模型得到的精度明显高于基于其它分类器的模型,而Bagging集成分类器使BAG-fold模型的预测能力增强预测精度提高。所以本文提出的方法能够提高模型预测性能得到更好的结果。
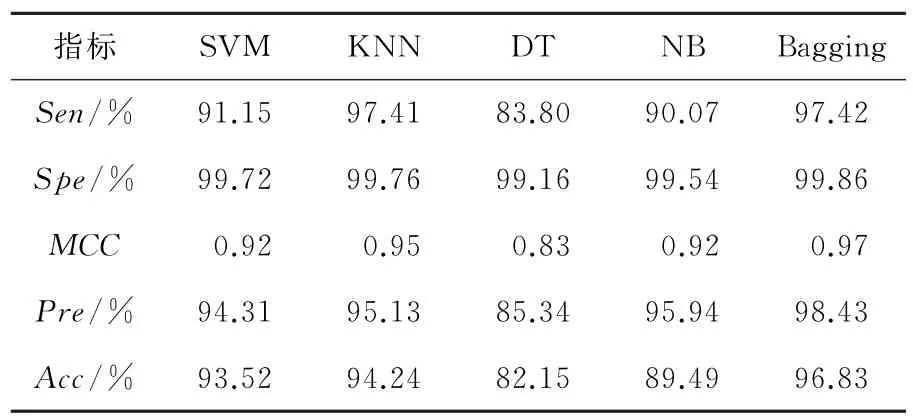
表8 Bagging集成方法与其他机器学习方法对比Table 8 Comparison of Bagging integration method and other machine learning methods
2.5 与其它方法进行比较
本工作在训练集DD 和测试集RDD 上的精度与其它论文中提到的方法Taxfold[33],SVMfold[6],TA-fold[6],MV-fold[4],MT-fold[4]进行了对比,如表9所示。
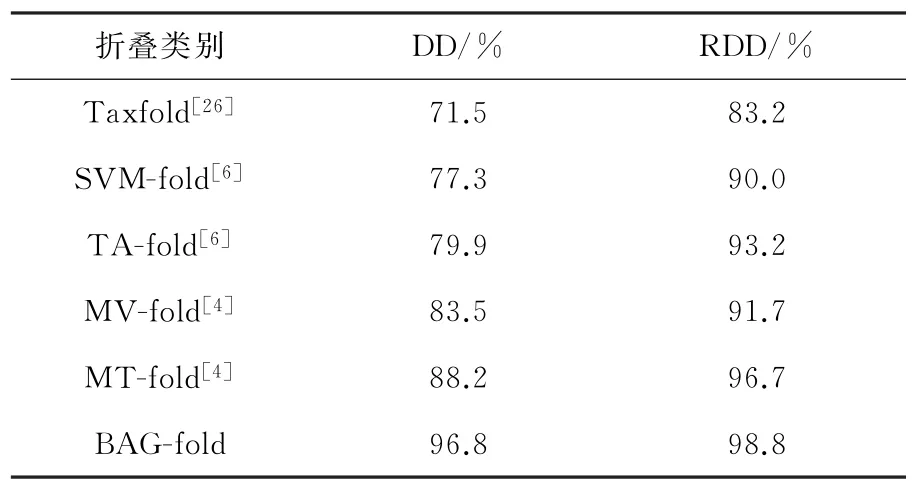
表9 BAG-fold方法与其它方法的比较Table 9 Comparison of BAG-fold method and other methods
由表9可知,DD 和RDD 数据集的Acc精度都达到了较高数值。本研究中DD 数据集和RDD 数据集的精度分别为96.8%和98.8%。在与最新论文中提到MV-fold和MT-fold的方法相比,在DD数据集上BAG-fold 模型比MV-fold 模型高出13.3%,而在RDD 数据集上BAG-fold 模型要比MV-fold模型高出7.1%。在DD 数据集上BAGfold模型比MT-fold模型高出8.6%,而在RDD 数据集上高出2.1%。通过比较,提出的BAG-fold模型在DD 和RDD 数据集上明显优于其它最新的蛋白质折叠识别模型。基于以上观察和比较,提出的方法优于现有模型。
3 结语
在折叠识别领域,传统的实验方法现已经无法满足科学研究的需要,大多数判别方法都是基于机器学习技术的,因此探索先进的机器学习方法以进一步提高预测性能至关重要。而集成分类器具有运行时间短,准确性高的优势,所以本研究的方法得到了较好的预测结果。本研究通过集成学习方法建立了BAG-fold预测模型。首先对DD 和RDD 数据集进行特征提取,与其它方法相比本研究对4种不同特征信息PsePSSM、DCCA、SS和EBGW 进行了融合。使用多种特征提取方法进行融合可以获得更多的蛋白质序列特征信息,可有效的防止单一特征提取方法对对蛋白质序列信息的损失。其次,采用LFDA 方法去除特征冗余信息选取最优特征子集。使用降维方法可以将融合多特征的蛋白质序列信息中的冗余信息进行去除,可减少对分类器性能的影响。最后,将最优特征子集输入到集成分类器中进行蛋白质折叠识别。使用10 折交叉验证的实验结果表明,所提出的方法能提高数据的精度。BAG-fold预测模型在DD和RDD数据集上的精度分别达到96.8%和98.8%。实验结果表明,对两个数据集进行评估,本研究所提出的BAGfold模型优于所有其它比较方法。尽管与其它方法相比,本研究的方法具有一定的的优越性,但还有进一步的提升空间。在今后的工作中将以提高模型预测精度为目的,在蛋白质折叠识别领域进行下一步研究。
