糖尿病心肌病发病机制的研究进展
2021-12-01廖敏杨洛王珍郝亚荣
廖敏,杨洛,王珍,郝亚荣
武汉大学人民医院老年病科,武汉430060
糖尿病是一种严重威胁人类健康的慢性代谢性疾病,其发病率呈现逐渐增加的趋势。2019年约有4.63亿人患有糖尿病,占全球成人人口的9.3%,预计这一数字将在2030年增加到5.78亿(10.2%),2045年增加到7亿(10.9%)[1]。随着发病率的增加,糖尿病的心血管并发症糖尿病心肌病(diabetic cardiomyopathy,DCM)逐渐受到临床医师的重视。1972年Rulber等[2]发现在没有明显的冠状动脉及心脏瓣膜病、高血压、酗酒和先天性心脏病的4例糖尿病肾小球硬化症患者中出现充血性心力衰竭和心脏肥大,首次提出DCM的概念。DCM是指在没有冠状动脉疾病、重大瓣膜病和其他常规心血管危险因素(如高血压和血脂异常)的糖尿病患者中出现心肌结构和功能障碍[3]。DCM病程可以分为四个阶段[4]:①射血分数正常的舒张功能障碍;②舒张功能障碍伴随收缩功能障碍;③舒张功能障碍和收缩功能障碍伴随微血管疾病或冠状动脉粥样硬化,无瓣膜疾病和高血压;④导致心力衰竭的临床明显缺血或梗死。DCM可导致50%以上糖尿病患者死亡,其特征是心脏脂质蓄积、心肌纤维化和心肌细胞死亡增加,均会导致左心室重构和肥大、舒张功能障碍,最终导致收缩功能障碍[5−6]。目前关于DCM的病理生理学机制尚未完全阐明,且临床缺乏特异性的治疗药物。本文从心肌细胞代谢紊乱、炎症反应和心肌纤维化三个方面总结了近年来国内外有关DCM病理生理机制的进展情况,旨在进一步加强对该疾病的认识和诊治。
1 心肌细胞代谢紊乱
心脏是一个高耗能器官,其主要供能物质是葡萄糖和脂肪酸。在生理状态下,长链脂肪酸β-氧化是心肌的首选能量底物。70%的能量消耗来自脂肪酸氧化,剩余的30%则来自葡萄糖、乳酸、酮类和氨基酸[7]。而糖尿病患者则会出现游离脂肪酸(free fatty acid,FFA)摄取增加和糖摄取减少等代谢紊乱。
1.1 高血糖和胰岛素抵抗(insulin resistance,IR)
IR是指胰岛素无法促进其在脂肪组织、骨骼肌和肝脏等器官中的代谢作用,表现为脂肪分解和肝脏葡萄糖生成的抑制作用受损,在肌肉和脂肪细胞中胰岛素介导的葡萄糖摄取障碍[8]。胰岛素除了维持细胞内稳态,还参与调节碳水化合物和脂类代谢,其分泌或功能失调可能会对心脏和平滑肌功能造成有害影响,包括血管活性激素生成和释放的失衡、内皮损伤和血管平滑肌细胞代谢缺陷。糖尿病中这些缺陷会导致心肌小动脉和毛细血管基底膜增厚(微血管病的早期标志),进一步加剧心肌纤维化和减少血流[9]。
CD36是一种介导FFA摄取的转运蛋白,葡萄糖转运体4(glucose transporter 4,GLUT4)是一种介导血浆中葡萄糖向心肌细胞膜转运并且广泛表达于心肌细胞的蛋白[10]。在2型糖尿病或IR状态下,CD36优先定位于肌细胞膜,而GLUT4返回细胞内[11]。在2型糖尿病动物模型中也观察到IR会削弱GLUT4在细胞膜表面的表达,导致糖摄取减少[12]。腺苷酸活化蛋白激酶是维持细胞摄取葡萄糖的生物能量代谢关键蛋白。研究发现,激活腺苷酸活化蛋白激酶磷酸化能够调控下游通路糖代谢调节蛋白,促进GLUT4的质膜转运,减轻DCM心肌细胞凋亡[13]。IR能够增加FFA的氧化水平,诱发心肌细胞生成大量活性氧,刺激过氧化物酶体增殖物激活受体α(peroxisome proliferator activated receptor α,PPAR-α)过表达和叉头盒子1核转位,导致心肌病[14]。另外,心脏中过量的脂质堆积也会产生一种“心脏脂毒性”,与心脏IR、心肌细胞凋亡和随后的收缩功能障碍有关[15]。IR与肾素-血管紧张素-醛固酮系统激活的相互作用可以导致DCM早期阶段的心脏纤维化和舒张功能障碍[11]。血管紧张素Ⅱ是肾素-血管紧张素系统的效应肽,糖尿病患者心脏容易受到血管紧张素Ⅱ的影响,导致心脏肥大、纤维化和炎症[16]。
IR和高血糖是DCM发生和发展的基础和核心。高血糖状态下,烟酰胺腺嘌呤二核苷酸磷酸氧化酶和蛋白激酶C激活、一氧化氮合酶解耦联、晚期糖基化终末产物(advanced glycation end products,AGEs)生成、线粒体电子传输链泄漏和线粒体通透性转换孔的形成,可以促进心脏活性氧合成增加,激活核因子-κB(nuclear factor-κB,NF-κB),导致心肌炎症、纤维化、肥大和氧化应激[17]。此外,AGEs通过刺激胶原表达、聚集和交联,导致心肌纤维化和顺应性降低[11]。总之,高血糖和IR诱导AGEs增加、心脏脂毒性、肾素-血管紧张素-醛固酮系统激活、微血管功能障碍、氧化应激和炎症反应,这些病理生理异常与心脏肥大、纤维化、舒张功能障碍和心力衰竭有关(图1)。
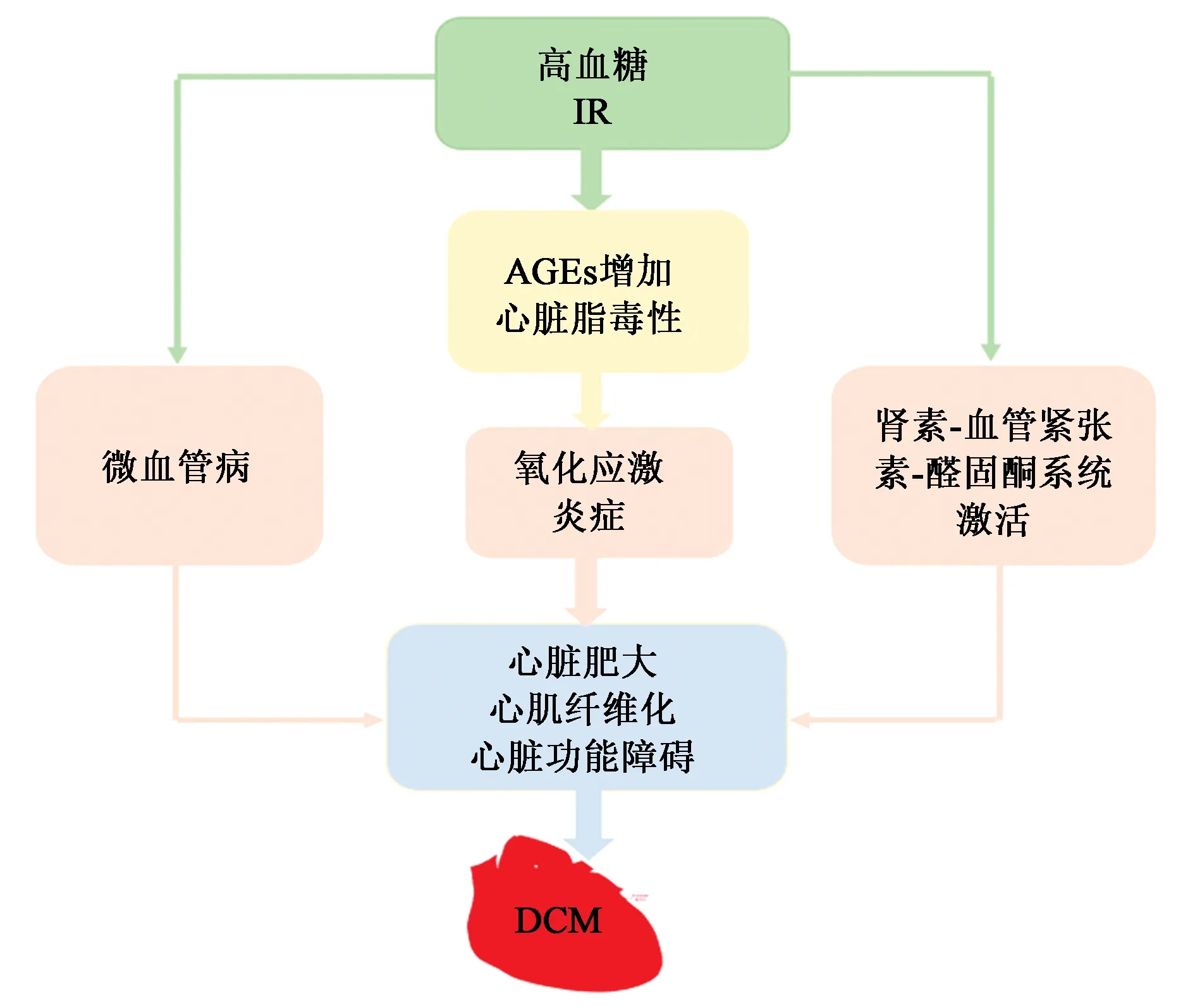
图1 糖尿病心肌病的发病机制Fig.1 Pathogenesis of diabetic cardiomyopathy
1.2 心脏脂毒性
IR和2型糖尿病患者肝细胞内脂质合成增加,脂肪细胞内脂质分解增加,导致循环FFA和甘油三酯水平升高。脂质积聚可直接阻碍心肌细胞代谢和收缩力,还可以通过增加内质网应激和活性氧的产生促进心肌细胞凋亡[11]。FFA诱导的脂毒性可以促进内质网应激、氧化应激、自噬、凋亡和炎症,从而导致组织损伤[18]。细胞内毒性代谢物三酰甘油和非必需脂肪酸(长链脂酰辅酶A、二酰甘油、神经酰胺)可以削弱胰岛素信号传导和葡萄糖代谢,引起严重IR[19]。糖尿病心脏中神经酰胺的增加会降低GLUT4的转运和胰岛素刺激的葡萄糖摄取[20]。心肌细胞中二酰甘油增加可通过激活蛋白激酶C而损害葡萄糖代谢,从而减少了胰岛素代谢的信号传导和一氧化氮的产生[20]。
PPAR-α是调节葡萄糖和脂质稳态的核受体,在心血管疾病中起关键作用[6]。糖尿病患者心脏中PPAR-α的表达增加,与脂肪酸摄取增加、甘油三酯积累和葡萄糖利用减少有关[20]。Liu等[5]发现横纹肌特异性E3连接酶mitsugumin53(MG53)是一种新型PPAR-α上游调节剂,在骨骼肌和心肌中大量表达。MG53过表达通过破坏胰岛素受体和胰岛素受体底物1、上调PPAR-α,导致IR和脂质蓄积,从而诱发DCM。此外,Wu等[21]证实高血糖或高胰岛素会诱导心脏分泌MG53,在2型糖尿病患者和动物模型中MG53水平升高,而且使用单克隆抗体中和MG53可以改善2型糖尿病db/db小鼠高血糖并增强胰岛素敏感性,为治疗2型糖尿病及其并发症开辟了新途径。
2型糖尿病患者的脂肪组织被单核细胞浸润,处于一种慢性炎症状态。脂肪细胞和巨噬细胞分泌促炎细胞因子或促血栓形成细胞因子,如肿 瘤 坏 死 因 子-α(tumour necrosis factor-α,TNF-α)、白介素-6(interleukin-6,IL-6)、抵抗素和血管紧张素原,导致动脉粥样硬化和IR[19]。糖尿病患者发生IR时,心肌细胞对葡萄糖利用率降低,主要依赖于FFA氧化供能,因此导致内质网应激、钙调节受损、线粒体功能障碍等,最终影响心肌舒张和收缩功能而导致心脏重构[22]。CD36即脂肪酸转位酶,是脂肪酸分解代谢中的限速酶,位于心肌细胞膜和胞质中,糖尿病时CD36从胞质转位到胞膜,摄取更多脂肪酸参与氧化供能[23]。研究发现,下调CD36的表达可以改善心肌纤维化,并减少心肌细胞凋亡[22]。
2 炎症反应
炎症反应是DCM发展过程的中心环节。FFA和内脏脂肪组织水平的提高促进全身和心血管炎症细胞因子的表达。全身和局部炎症可能导致心肌结构和代谢的改变,可进展为舒张功能障碍,最终导致心力衰竭[11]。在肥胖和IR状态下,M1型巨噬细胞分泌炎症细胞因子,导致全身和心脏胰岛素信号传导降低,并促进DCM的发展[11]。T淋巴细胞促进趋化因子、促炎细胞因子和生长因子的分泌,进而导致心肌纤维化和舒张功能障碍[24]。
高糖刺激下增强糖基化,导致AGEs和脂质与AGEs受体结合,产生活性氧,激活NF-κB,导致促炎介质TNF-α和IL-1β生成[17]。核苷酸结合寡聚化结构域样受体蛋白3(nucleotide-binding oligomerization domain-like receptor protein 3,NLRP3)炎性小体是一种多蛋白复合物,在炎症过程中发挥重要作用。硫氧还蛋白结合蛋白(thioredoxin-interacting protein,TXNIP)是高血糖诱导的早期炎症介质,具有激活NLRP3炎性小体的能力。抑制TXNIP-NLRP3的活化可以抑制高糖诱导的氧化应激和细胞外基质的蓄积[25]。因此,活性氧生成增加会导致炎性介质的增加,同时,炎性介质的增多可以刺激活性氧的生成,形成恶性循环。TGR5是一种G蛋白偶联受体,Deng等[26]证实通过抑制NF-κB和核因子E2相关因子2激活TGR5,能够减轻高糖诱导的心肌细胞炎症和氧化应激。因此,TGR5被认为是治疗DCM的潜在药理靶点。在体内外研究中,过量的饱和脂肪酸棕榈酸与髓样分化蛋白2(myeloid differentiation protein 2,MD2)直接结合激活Toll样受体4,诱导TNF-α、IL-6、单核细胞趋化蛋白-1和细胞内粘附分子-1的生成,敲除MD2可以减轻心肌组织促炎介质的产生[27]。MD2基因沉默可以减轻高糖诱导的大鼠心肌细胞炎症和凋亡,可能与负反馈调控细胞外信号调节激酶、P38丝裂原活化蛋白激酶和C-Jun氨基末端激酶有关[28]。促炎细胞因子和趋化因子可能在心脏间质中募集纤维源性白细胞亚群[29]。Zhang等[30]发现炎症细胞因子IL-6在DCM小鼠血清和心脏中的表达增加,可上调转化 生 长 因 子-β1(transforming growth factor-β1,TGF-β1)的表达,从而促进Ⅰ型胶原和Ⅲ型胶原蛋白的表达,最终导致心脏功能受损和纤维化。
3 心肌纤维化
心肌纤维化是DCM和最终导致心力衰竭的一个重要原因。心肌纤维化是高血糖诱导的细胞外基质重构加重的结果,其特征是细胞外基质沉积增加、胶原溶解受损、心肌成纤维细胞增殖和心肌僵硬,伴随心功能障碍[31]。TGF-β1是介导纤维化相关信号通路的重要因子,在DCM心肌纤维化中发挥重要作用。Ⅰ型胶原和Ⅲ型胶原是细胞外基质的主要成分,当心肌胶原比例失衡时,心室壁僵硬度下降,心室舒张改变,最终导致心衰,构成了心肌纤维化的主要病理基础[32]。TGF-β1刺激可显著增加Ⅰ型胶原和Ⅲ型胶原的表达[33]。高糖刺激TGF-β1活化,其下游Smad2和Smad3蛋白被激活并磷酸化,从而参与组织和器官的纤维化。抑制TGF-β1和p-Smad2/3的表达,可以减轻糖尿病心肌纤维化和胶原沉积[34]。Gbr等[35]发现激活PPAR-γ和抑制钙调素依赖蛋白激酶Ⅱ/NF-κB/TGF-β1,可以减轻糖尿病大鼠的氧化应激、炎症和纤维化。研究报道,激活PPAR-γ可以减轻DCM心肌纤维化,与调节TGF-β/细胞外调节蛋白激酶通路和上皮-间质转化有关[36]。基质金属蛋白酶调控细胞外基质的合成和降解,Li等[37]证实糖尿病大鼠心肌中基质金属蛋白酶2和基质金属蛋白酶9的表达增加,促进心肌成纤维细胞分化和胶原生成,导致心肌功能紊乱和纤维化。也有研究报道,在链脲佐菌素诱导的糖尿病小鼠心脏中基质金属蛋白酶2的表达下降,胶原降解减少,从而促进了心脏纤维化[38]。糖尿病患者心脏中基质金属蛋白酶的表达出现相反结果,可能与机体的代偿机制有关,但还需要进一步研究证实。
Adebiyi等[31]证实,通过抑制烟酰胺腺嘌呤二核苷酸磷酸氧化酶活性以及下调蛋白激酶C和p38丝裂原活化蛋白激酶,可减轻氧化应激,从而延缓糖尿病心肌纤维化。趋化因子也可能通过募集成纤维单核细胞亚群或通过直接作用于成纤维细胞而诱导心脏纤维化[29]。TNF-α刺激诱导心肌成纤维细胞增殖和增加胶原合成。另一方面,IL-1β延迟肌成纤维细胞转化,促进α基质降解促炎性成纤维细胞[29]。研究发现,诱导M2型巨噬细胞极化,减少M1型巨噬细胞极化和炎症反应可以减轻DCM小鼠的心肌纤维化[39]。
4 展望
DCM是糖尿病常见且严重的心血管并发症之一,严重影响患者的生活质量和生命安全,也是糖尿病相关发病率和死亡率居高不下的主要原因。DCM的病理生理机制复杂,而心肌细胞代谢紊乱、心肌纤维化、炎症反应在DCM的进展中发挥至关重要的作用。从目前的研究中可以发现,针对这些病理机制的治疗可能会使DCM患者受益。然而,由于目前对于DCM的研究仅局限于动物和细胞模型,因此对其发病机制的认识仍然有限,DCM的发病机理和潜在的治疗靶点仍然需要进一步深入探索。
