枫泾猪、梅山猪和沙乌头猪全基因组ROH检测及候选基因鉴定
2021-11-14赵旭庭徐盼马政仲德刘林雨倪黎纲吴井生
赵旭庭 徐盼 马政 仲德 刘林雨 倪黎纲 吴井生


摘要: 通過检测枫泾猪、梅山猪和沙乌头猪的长纯合片段(Runs of homozygosity, ROH),鉴定其与3个猪种经济性状相关的候选基因。利用全基因组单核苷酸多态性(Single Nucleotide Polymorphisms, SNP)芯片数据对这3个猪群体进行全基因组ROH检测,统计ROH数量、长度及分布,计算基于ROH的近交系数( F ROH ),并在高频ROH区域鉴定候选功能基因。结果显示,从枫泾猪、梅山猪和沙乌头猪群体中分别检测到 1 588 个、 3 314 个和 4 419 个ROH。枫泾猪的ROH平均长度为12.84 Mb,平均 F ROH 为0.084;梅山猪的ROH平均长度为10.55 Mb,平均 F ROH 为0.164;沙乌头猪的ROH平均长度为10.52 Mb,平均 F ROH 为0.141。在高频ROH区域共鉴定到9个枫泾猪候选功能基因和8个梅山猪候选功能基因。研究结果为枫泾猪、梅山猪和沙乌头猪的资源保护和分子标记辅助选择奠定了基础。
关键词: 猪; 长纯合片段; 近交系数; 候选基因
中图分类号: S813.1 文献标识码: A 文章编号: 1000-4440(2021)05-1234-10
Genome-wide scan for run of homozygosity and identification of corresponding candidate genes in Fengjing pigs, Meishan pigs and Shawutou pigs
ZHAO Xu-ting 1 , XU Pan 1 , MA Zheng 1 , ZHONG De 1 , LIU Lin-yu 1 , NI Li-gang 1 , WU Jing-sheng 2
(1.Jiangsu Agri-animal Husbandry Vocational College, Taizhou 225300, China; 2.Jiangsu Vocational College of Agriculture and Forestry, Jurong 212400, China)
Abstract: By detecting runs of homozygosity (ROH) of Fengjing pig, Meishan pig and Shawutou pig, the candidate genes related to the economic traits of the three pig breeds were identified. The genome-wide single nucleotide polymorphisms (SNP) chip data were used to perform genome-wide ROH scan for the three pig breeds. The number, length and distribution of ROH were counted, and the inbreeding coefficient ( F ROH ) was calculated based on ROH. In addition, the functional candidate genes were identified in the high frequency ROH region. The results showed that 1 588 , 3 314 and 4 419 ROH were detected in the three pig populations, respectively. The average length of ROH was 12.84 Mb and the average F ROH was 0.084 in Fengjing pigs. The average length of ROH was 10.55 Mb and the average F ROH was 0.164 in Meishan pigs. The average length of ROH was 10.52 Mb and the average F ROH was 0.141 in Shawutou pigs. Nine functional candidate genes of Fengjing pig and eight functional candidate genes of Meishan pig were identified in high frequency ROH region. The results lay a foundation for resource conservation and molecular marker-assisted selection of Fengjing pig, Meishan pig and Shawutou pig.
Key words: pig; runs of homozygosity (ROH); inbreeding coefficient; candidate gene
长纯合片段(Runs of homozygosity, ROH)是基因组上一定数量、一定密度的单核苷酸多态性(Single nucleotide ploymorhphisms, SNPs)表现为纯合的一段区域 [1] 。在人工选择的过程中,畜禽群体的基因组被重塑,所选区域中的ROH频率会提高,甚至产生“ROH岛”。ROH信息在畜禽亲缘关系鉴定、遗传多样性分析、近交衰退评估、选择信号鉴定和功能基因筛选等方面发挥着重要作用。Keller等 [2] 在比较基于ROH的近交系数( F ROH )、基于基因组杂合度的近交系数( F HOM )、基于基因组关系矩阵的近交系数( F GRM )和基于系谱信息的近交系数( F PED )后发现, F ROH 是最有效的估计近交的方法。Mastrangelo等 [3] 在Pontremolese、Varzese-Ottonese和Mucca Pisana群体中检测到高水平的ROH,针对这些群体,在实施配种计划中应尽量增加种公畜血统,减少其遗传多样性的损失,维持或者增加其群体有效含量。Saura等 [4] 分析了近交对伊比利亚猪繁殖性状的影响,结果表明,近交系数每增加0.1,其仔猪初生后的存活率、仔猪出生后的总数量有下降趋势。Zhong等 [5] 利用简化基因组测序数据在金华猪群体的ROH岛中鉴别到与繁殖、肉质、食欲及抗病性状相关的基因。
枫泾猪、梅山猪和沙乌头猪是江苏省优良的地方猪遗传资源,以产仔数高、肉质优良、耐粗饲和环境适应力强等特点而闻名。本研究利用中芯一号育种芯片数据对这3个猪群体进行全基因组ROH检测,统计ROH数量、长度及分布,计算基于ROH的近交系数,并在高频ROH区域鉴定候选功能基因,以期为枫泾猪、梅山猪和沙乌头猪的资源保护和分子标记辅助选择奠定基础。
1 材料与方法
1.1 试验动物
试验动物为来自镇江牧苑动物科技开发有限公司省级枫泾猪保种场的107头枫泾猪(FJ)、江苏农林职业技术学院国家级梅山猪保种场的94头梅山猪(MS)和江苏兴旺农牧科技发展有限公司国家级沙乌头猪保种场的146头沙乌头猪(SWT)。采集猪耳组织样后保存于 -20 ℃ 冰箱内待用。
1.2 SNP芯片分型及质量控制
委托北京康普森生物技术有限公司使用中芯一号育种芯片对107头枫泾猪、94头梅山猪和146头沙乌头猪进行基因分型。用plink软件分别对3个猪群体的芯片数据进行质量控制,质量控制标准:1)个体检出 率≥ 90%;2)SNP检出 率≥ 90%;3)次等位基因频率( MAF )≥0.01;4)哈迪温伯格平衡 P ≥ 1× 10 -5 ;5)剔除性染色体。
1.3 全基因组ROH检测
ROH检测使用Plink软件,使用滑动窗口的方法对常染色体进行检测,具体的检测参数:1)ROH长度大于1 Mb;2)至少连续50个SNPs;3)连續SNP间的距离小于1 Mb;4)最多允许ROH中有5个缺失和1个杂合;5)窗口阈值为0.01;6)密度为0.01 SNP/kb 。
1.4 基因组近交系数的计算
利用ROH计算近交系数( F ROH ),公式如下:
F ROH =∑ L ROH / L auto
其中:∑ L ROH 为常染色体上ROH片段的长度之和, L auto 为常染色体的物理总长度。
根据上式计算 F ROH 1.0~5.0 Mb 、 F ROH 5.1~10.0 Mb 、 F ROH>10.0 Mb 和 F ROH total 4种近交系数之间的Pearson相关系数,其中 F ROH total 为基于全部长度的ROH计算的近交系数。
1.5 高频ROH区域的检测及候选基因的鉴定
统计每个SNP在各自猪群体内参与组成ROH的次数占样本数的比例,并将高于45%的SNP区域作为高频ROH区域,利用RCircos包绘制3个猪群体ROH频率在染色体上分布的Circos图。基于高频ROH区段的物理位置,在猪基因组注释文件(ftp://ftp.ensembl.org/pub/release-100/gtf/sus_scrofa/Sus_scrofa.Sscrofa11.1.100.chr.gtf.gz)中检索基因。
利用DAVID数据库(https://david.ncifcrf.gov/)对高频ROH区域注释到的基因进行GO(Gene ontology,基因本体)功能富集和KEGG(Kyoto Encyclopedia of Genes and Genomes,京都基因与基因组百科全书)Pathway富集分析,当 P ≤ 0.05时,则表示显著富集。将显著富集到生物学过程和信号通路的基因与猪数量性状基因座(Quantitative Trait Locus, QTL)数据库(https://www.animalgenome.org/cgi-bin/QTLdb/SS/index)进行比对,获得候选功能基因。
2 结果与分析
2.1 基因型的质量控制
中芯一号育种芯片含有 51 315 个SNP位点,经过质量控制后,对剩余107头枫泾猪的 19 895 个SNPs、94头梅山猪的 24 526 个SNPs和146头沙乌头猪的 32 275 个SNPs进行后续分析。
2.2 ROH数量、长度及分布统计
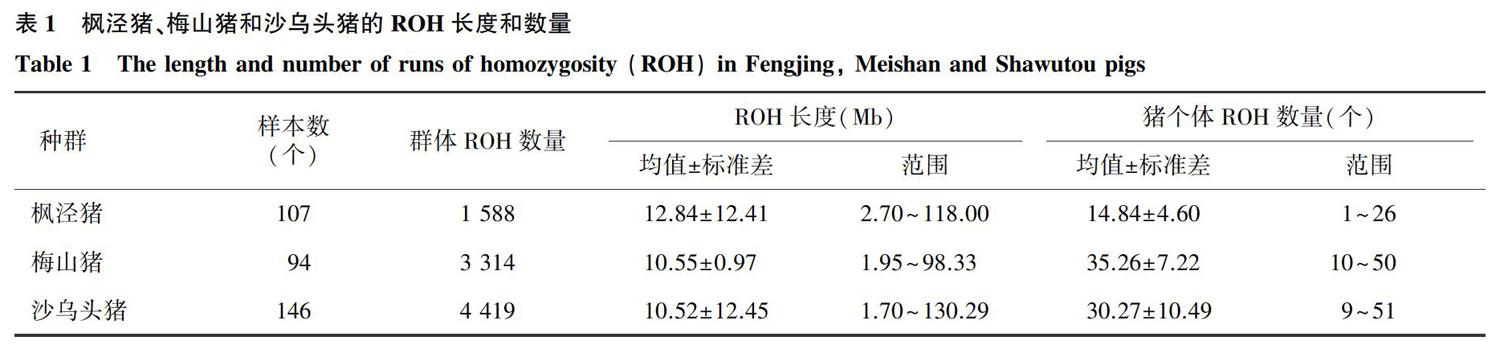
由表1、图1、图2可以看出,从枫泾猪、梅山猪和沙乌头猪群体中分别检测到 1 588 个、 3 314 个和 4 419 个ROH,其长度主要集中在40.00 Mb以内。枫泾猪ROH的平均长度为12.84 Mb;ROH的最长长度为118.00 Mb,含有 1 328 个SNPs,位于第15号染色体上;ROH的最短长度为2.70 Mb,含有50个SNPs,位于第13号染色体上;枫泾猪个体的平均ROH数量为14.84个,以15号染色体上的ROH最多,18号染色体上的ROH最少。梅山猪ROH的平均长度为10.55 Mb;ROH的最长长度为98.33 Mb,含有 1 313 个SNPs,位于第14号染色体上;ROH的最短长度为1.95 Mb,含有59个SNPs,位于第6号染色体上;梅山猪个体ROH的平均数量为35.26个,14号染色体上的ROH最多,12号染色体上的ROH最少。沙乌头猪ROH的平均长度为10.52 Mb;ROH的最长长度为130.29 Mb,含有 1 727 个SNPs,位于第1号染色体上;ROH的最短长度为1.70 Mb,含有50个SNPs,位于第6号染色体上;沙乌头猪个体ROH的平均数量为30.27个,1号染色体上的ROH最多,10号染色体上的ROH最少。
2.3 近交系数
由表2可以看出,枫泾猪基因组 F ROH total 的最大值为0.171,最小值为0.003,平均值为0.084;梅山猪基因组 F ROH total 的最大值为0.305,最小值为0.057,平均值为0.164;沙乌头猪基因组 F ROH total 的最大值为0.501,最小值为0.022,平均值为0.141;3个群体中梅山猪的平均 F ROH total 最高。对比表2中3种猪群体的4种近交系数发现,枫泾猪群体中 F ROH 5.1~10.0 Mb 与 F ROH >10.0 Mb 、 F ROH total 分别在0.05、 0.001 水平显著相關, F ROH >10.0 Mb 与 F ROH total 在0.001水平显著相关;梅山猪群体中 F ROH 5.1~10.0 Mb 与 F ROH 1.0~5.0 Mb 、 F ROH total 及 F ROH >10.0 Mb 与 F ROH total 均在0.001水平显著相关;沙乌头猪群体中 F ROH 1.0~5.0 Mb 与 F ROH total 在0.01水平显著相关, F ROH 1.0~5.0 Mb 与 F ROH 5.1~10.0 Mb 、 F ROH 5.1~10.0 Mb 与 F ROH >10.0 Mb 、 F ROH 5.1~10.0 Mb 与 F ROH total 、 F ROH >10.0 Mb 与 F ROH total 均在0.001水平显著相关(表3)。
2.4 高频ROH区域及候选基因的鉴定
本研究选取45%作为高频ROH区域的阈值,在枫泾猪、梅山猪和沙乌头猪群体中分别检测到7个、20个和9个高频ROH区域,分别注释到414个、547个和151个基因(表4)。枫泾猪、梅山猪和沙乌头猪没有共同的高频ROH区域,枫泾猪和梅山猪在第14号染色体上有共同高频ROH区域,梅山猪、沙乌头猪在第8号、11号染色体上有共同高频ROH区域(图3)。
利用DAVID数据库分别对枫泾猪、梅山猪和沙乌头猪群体中注释到的414个、547个和151个基因进行GO功能富集分析和KEGG Pathway富集分析,结果分别见表5、表6和表7。分析得出,枫泾猪群体的414个基因中的136个基因显著富集于谷胱甘肽代谢过程、肝脏发育、炎症反应的负调控等22个GO条目和朊病毒病等4个信号通路;梅山猪群体的547个基因中的134个基因显著富集于半胱氨酸型内肽酶活性的负调节、谷胱甘肽代谢过程、类黄酮葡萄糖醛酸化等22个GO条目和11个信号通路;沙乌头猪群体的151个基因中的24个基因显著富集于类黄酮葡萄糖醛酸化、类黄酮生物合成过程、生物矿物组织发育等11个GO条目和抗坏血酸和醛糖酸盐代谢等9个信号通路。分别将枫泾猪群体的136个基因、梅山猪群体的134个基因和沙乌头猪群体的24个基因与猪QTL数据库进行比对,得到枫泾猪候选功能基因 HNF1A、CDH17、LIF、DECR1、ARL15、ACTN2、FABP5、FABP4和EWSR1 (表5),梅山猪候选功能基因 CTSH、MSTN、GC、VEGFA、ZDHHC2、MTMR7、CDC5L和LIF (表6)。
3 讨 论
ROH可以指示亲缘的远近和近交的历史,ROH片段越长,家系内存在近世代近交的可能性越高 [2, 6] 。在0~5.0 Mb长度的ROH中,枫泾猪所占比例最低,沙乌头猪所占比例最高;在大于5.0 Mb长度的ROH中,枫泾猪所占比例最高,沙乌头猪所占比例最低。该结果与ROH平均长度的结果一致(枫泾猪ROH的平均长度最长,沙乌头猪ROH的平均长度最短)。3个猪群中长度大于5.0 Mb的ROH所占比例都较高,表明3个猪群经历了较高程度的近交,可能与3个猪群的群体规模有关。在过去几十年里,欧洲现代品种猪(主要是大白猪、长白猪和杜洛克猪)因瘦肉率高、生长速度快、饲料转化率高而不断被引进中国,目前占据着中国养猪业的统治地位,导致中国地方猪的养殖数量显著减少,枫泾猪、梅山猪和沙乌头猪的种群规模也因此受到影响。在本研究中,枫泾猪、梅山猪和沙乌头猪样品采集于保种场,种群数量有限,从而导致近世代较高程度的近交。
每個环代表1个猪群体,从内到外的环分别为枫泾猪、梅山猪、沙乌头猪,柱子代表ROH频率。图中数据表示染色体的位置信息。
研究发现,基于ROH的近交系数( F ROH )与基于系谱信息的近交系数( F PED )间的相关系数随着ROH长度的增加而提高,在群体系谱信息不全的情况下, F ROH 可以替代 F PED 来评估近交程度 [7] 。枫泾猪、梅山猪和沙乌头猪保种场早期的猪主要从主产区及周边长期从事地方猪饲养的农户收集而来,系谱信息不全,因此需要通过基于ROH的近交系数( F ROH )来评估其近交程度。本研究基于ROH计算了每个猪群的4种近交系数( F ROH 1.0~5.0 Mb 、 F ROH 5.1~10.0 Mb 、 F ROH >10.0 Mb 和 F ROH total ),结果表明,3个猪群中枫泾猪的近交程度最低,梅山猪的近交程度最高。与 F ROH 1.0~5.0 Mb 、 F ROH 5.1~10.0 Mb 相比,3个猪群的 F ROH >10.0 Mb 与 F ROH total 最接近,与4种近交系数间的相关性分析结果一致, F ROH >10.0 Mb 和 F ROH total 间的相关系数最高。Zhong等 [5] 利用简化基因组测序数据计算了金华猪的 F ROH 1.0~5.0 Mb 、 F ROH 5.1~10.0 Mb 、 F ROH >10.0 Mb 和 F ROH all ,结果表明, F ROH >10.0 Mb 和 F ROH all 间的相关系数最高,与本研究结果一致。
本研究通过将富集到生物学过程和信号通路的基因与猪QTL数据库进行比对,鉴定到9个枫泾猪候选功能基因和8个梅山猪候选功能基因,其中 LIF 基因是共同的候选功能基因, HNF1A 基因是猪肉质和胴体性状的候选基因 [8] , CDH17 基因影响公猪睾丸中雄烯酮水平 [9] , LIF 基因影响猪的产活仔数和21日龄仔猪数 [10] , DECR1 基因与杜洛克猪脂质组成性状显著相关 [11] , ARL15 基因是猪的眼肌面积的候选基因 [12] , ACTN2 基因与猪肉的烹饪损失有关 [13] , FABP5 和 FABP4 基因与猪的脂肪性状相关 [14-15] , EWSR1 基因影响猪肉肉色 [13] , CTSH 基因与饲料增质量比相关 [16] , MSTN 基因与猪日增质量和体质量等性状显著相关 [17] , GC 基因与皮特兰猪的pH 24 h 相关 [18] , VEGFA 基因影响猪后腿步态评分 [19] , ZDHHC2 基因是鲁莱黑猪肌内脂肪含量的候选基因 [20] , MTMR7 基因与猪血液中平均红细胞血红蛋白含量有关 [21] , CDC5L 基因与猪血细胞比容和血红蛋白相关 [22] 。本研究鉴定到的候选功能基因与猪的生长发育、胴体性状、肉质性状和繁殖性状相关,下一步试验将利用候选基因法研究候选功能基因与相关性状的关系,从而为解析猪生长发育、胴体性状、肉质性状和繁殖性状的分子机制提供参考。
参考文献:
[1] GIBSON J, MORTON N E, COLLINS A. Extended tracts of homozygosity in outbred human populations[J]. Human Molecular Genetics, 2006, 15(5): 789-795.
[2] KELLER M C, VISSCHER P M, GODDARD M E. Quantification of inbreeding due to distant ancestors and its detection using dense single nucleotide polymorphism data[J]. Genetics, 2011, 189(1): 237-249.
[3] MASTRANGELO S, SARDINA M T, TOLONE M, et al. Genome-wide identification of runs of homozygosity islands and associated genes in local dairy cattle breeds[J]. Animal, 2018, 12(12): 2480-2488.
[4] SAURA M, FERNNDEZ A, VARONA L, et al. Detecting inbreeding depression for reproductive traits in Iberian pigs using genome-wide data[J]. Genetics Selection Evolution, 2015, 47(1): 1.
[5] ZHONG X, SUN H, ZHANG Z, et al. Assessment of autozygosity derived from runs of homozygosity in Jinhua pigs disclosed by sequencing data[J]. Frontiers in Genetics, 2019, 10: 274.
[6] HOWRIGAN D P, SIMONSON M A, KELLER M C. Detecting autozygosity through runs of homozygosity: a comparison of three autozygosity detection algorithms[J]. BMC Genomics, 2011, 12(1): 460.
[7] PERIPOLLI E, STAFUZZA N B, MUNARI D P, et al. Assessment of runs of homozygosity islands and estimates of genomic inbreeding in Gyr ( Bos indicus ) dairy cattle[J]. BMC Genomics, 2018, 19(1): 34.
[8] KAYAN A, UDDIN M J, KOCAMIS H, et al. Association and expression analysis of porcine HNF1A gene related to meat and carcass quality traits[J]. Meat Science, 2013, 94(4): 474-479.
[9] DRAG M H, KOGELMAN L J, MARIBO H, et al. Characterization of eQTLs associated with androstenone by RNA sequencing in porcine testis[J]. Physiological Genomics, 2019, 51(10): 488-499.
[10] MUCHA A, ROPKA-MOLIK K, PIRKOWSKA K, et al. Effect of EGF, AREG and LIF genes polymorphisms on reproductive traits in pigs[J]. Animal Reproduction Science, 2013, 137(1/2): 88-92.
[11] RAMREZ O, QUINTANILLA R, VARONA L, et al. DECR1 and ME1 genotypes are associated with lipid composition traits in Duroc pigs[J]. Journal of Animal Breeding nad Genetics, 2014, 131(1): 46-52.
[12] ZHUANG Z W, LI S Y, DING R R, et al. Meta-analysis of genome-wide association studies for loin muscle area and loin muscle depth in two Duroc pig populations[J]. PLoS One, 2019, 14(6): e0218263.
[13] LI X P, KIM S W, DO K T, et al. Analyses of porcine public SNPs in coding-gene regions by re-sequencing and phenotypic association studies[J]. Molecular Biology Reports, 2011, 38(6): 3805-3820.
[14] ESTELL J, PREZ-ENCISO M, MERCAD A, et al. Characterization of the porcine FABP5 gene and its association with the FAT1 QTL in an Iberian by Landrace cross[J]. Animal Genetics, 2006, 37(6): 589-591.
[15] MERCAD A, PREZ-ENCISO M, VARONA L, et al. Adipocyte fatty-acid binding protein is closely associated to the porcine locus on chromosome 4[J]. Journal of Animal Science, 2006, 84(11): 2907-2913.
[16] RUSSO V, FONTANESI L, SCOTTI E, et al. Single nucleotide polymorphisms in several porcine cathepsin genes are associated with growth, carcass, and production traits in Italian Large White pigs[J]. Journal of Animal Science, 2008, 86(12): 3300-3314.
[17] TU P A, SHIAU J W, DING S T, et al. The association of genetic variations in the promoter region of myostatin gene with growth traits in Duroc pigs[J]. Animal Biotechnology, 2012, 23(4): 291-298.
[18] CINAR M U, KAYAN A, UDDIN M J, et al. Association and expression quantitative trait loci (eQTL) analysis of porcine AMBP, GC and PPP1R3B genes with meat quality traits[J]. Molecular Biology Reports, 2012, 39(4): 4809-4821.
[19] GUO Y M, ZHANG X F, REN J, et al. A joint genomewide association analysis of pig leg weakness and its related traits in an F 2 population and a Sutai population[J]. Journal of Animal Science, 2013, 91(9): 4060-4068.
[20] WANG Y P, NING C, WANG C, et al. Genome-wide association study for intramuscular fat content in Chinese Lulai black pigs[J]. Asian Australasian Journal of Animal Sciences, 2019, 32(5): 607-613.
[21] WANG J Y, LUO Y R, FU W X, et al. Genome-wide association studies for hematological traits in swine[J]. Animal Genetics, 2013, 44(1): 34-43.
[22] ZHANG Z Y, HONG Y, GAO J, et al. Genome-wide association study reveals constant and specific loci for hematological traits at three time stages in a White Duroc×Erhualian F 2 resource population[J]. PLoS One, 2013, 8(5): e63665.
(責任编辑:徐 艳)
