钛硅分子筛TS-1催化环氧丙烷异构反应的机理探究
2021-10-31王刚段学志袁渭康周兴贵
王刚,段学志,袁渭康,周兴贵
(华东理工大学化学工程联合国家重点实验室,上海 200237)
引 言
环氧丙烷(PO)是重要的有机化工原料,主要用于合成聚醚多元醇进而生产聚氨酯[1-2]。相比于工业上PO的生产方法,丙烯氢氧环氧化一步制备PO具有过程简单、绿色环保、原子利用率高等优势[3-7]。大量的含钛材料被用于该反应,其中,钛硅分子筛TS-1负载的纳米金催化剂(Au/TS-1)因其优异的PO性能受到广泛关注[8-12]。Zhou等[13-14]研究发现Au/TS-1催化剂快速失活的主要原因是结焦导致的微孔堵塞,提出了采用未焙烧的堵孔钛硅分子筛负载纳米金颗粒(Au/TS-1-B),结果表明可显著提高催化剂的稳定性。值得指出的是,除了主产物PO外,反应过程中往往有丙醛等副产物的生成。通过对该催化剂上副产物的分布进行分析可发现,丙醛和丙酮的选择性明显高于其他副产物,且在反应过程中呈现逐渐上升的趋势[15-16]。这些副产物的形成降低了PO的选择性和催化剂长周期稳定性,为丙烯氢氧环氧化催化剂的设计与优化带来了挑战。Delgass等[17]对该反应进行了H2/D2同位素实验,发现当原料气由H2切换为D2后,除了丙烯醛外,各产物的生成速率均下降,并提出这些副产物可能是由PO发生深度反应转化而来。Haruta等[18]通过动力学研究发现:随着丙烯转化率的增加,PO收率先上升后下降,而副产物的收率急剧上升,这预示着丙醛和丙酮可能是PO异构化的产物。Mul等[19]通过红外光谱证实PO可转化成双配位丙氧基物种,并认为该中间体可进一步转化为丙醛和丙酮副产物。对于PO在分子筛上的异构过程,Limtrakul等[20]采用理论计算研究了PO在ZSM-5分子筛酸性位上的异构机制,提出PO的异构主要存在两个过渡态:碳氧环的质子化及断裂;碳上氢原子的转移重排。但目前对于PO在钛硅分子筛上的异构机制尚未见报道,亟需对该过程进行机理性的认识与理解(图1)。
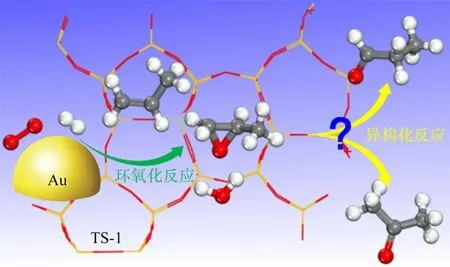
图1 Au/TS-1催化丙烯氢氧环氧化生成PO以及PO异构化示意图Fig.1 Schematic diagram of propylene epoxidation with H2 and O2 to PO and its isomerization over Au/TS-1 catalyst
本文首先制备了堵孔TS-1分子筛和Au/TS-1-B催化剂,并在固定床反应器中考察了其催化PO转化的性能,进一步采用DFT理论计算研究了PO分子在TS-1的Ti-Defect位点附近转化生成丙醛和丙酮的吸附态以及过渡态,提出了TS-1催化PO异构化的反应机理。这些研究结果将为丙烯氢氧环氧化催化剂上PO选择性的进一步调控与优化提供理论依据。
1 实验材料和方法
1.1 TS-1-B分子筛和Au/TS-1-B催化剂的制备
堵孔钛硅分子筛的制备按照文献中报道的水热合成方法进行[9,13],具体步骤如下:在42℃下将2 g吐温20溶于28 g超纯水中,并加入质量分数为25%的四丙基氢氧化铵水溶液,搅拌过程中向溶液中加入40.5 g正硅酸乙酯。然后将0.67 g钛酸四丁酯溶于15 g异丙醇溶液并添加到上述溶液中,利用水浴锅将溶液升温至80℃进行除醇。随后将溶液转移至水热釜中并在170℃下晶化48 h。将产物进行离心和洗涤,并在100℃下干燥12 h得到白色固体,即为堵孔钛硅分子筛,标记为TS-1-B。
Au/TS-1-B催化剂的制备参照文献中报道的尿素沉积沉淀法(DPU)进行[17]。首先将1 g上述TS-1-B加入40 ml超纯水中并搅拌,加入一定量的氯金酸的水溶液和尿素。随后将该悬浊液升温至90℃并保持6 h。随后将固体和液体采用离心分离,用40 ml超纯水洗涤,将获得的固体在室温下真空干燥得到未还原的金催化剂样品。
1.2 样品表征技术
采用X射线衍射(XRD)技术日本理学电机公司,18KW/D/MAX 2550 VB/PC对TS-1-B的物相结构进行分析,管电压和管电流分别为40 kV和450 mA。
TS-1-B中Ti的配位状态由紫外可见光谱(UVvis)美国Varian公司,Lambda 950型号紫外-可见-近红外分光光度计进行分析。以硫酸钡的光谱作为背景并扣除,仪器的波长范围为175~3300 nm,UV-Vis波段的精度为±0.1 nm。
采用高分辨率透射电子显微镜(HRTEM),日本JEOL公司,JEM-2100型透射电子显微镜对分子筛的形貌和大小进行分析,其点和线分辨率分别为0.23 nm和0.14 nm。
采用电感耦合等离子原子发射光谱(ICPAES),美国安捷伦公司,Varian 710 ES型原子吸收光谱仪测定所合成的催化剂中Au和Ti的含量。测试之前先采用HF溶液和王水将催化剂溶解,然后进行测定分析,结合标准曲线和堵孔钛硅分子筛中模板剂的含量约为16%[15]的结果,测得制备的Au/TS-1-B催化剂上Au元素的质量分数约为0.1%,分子筛中硅钛比约为93。
催化剂上纳米金颗粒的粒径分布使用中国科学院苏州纳米所的高角环形暗场-扫描透射电子显微镜(HAADF-STEM)美国FEI公司Tecnai G2 F20 S-TWIN透射电子显微镜进行确定。
使用傅里叶变换红外光谱(FT-IR)探究PO在TS-1-B分子筛和Au/TS-1-B催化剂上的吸附作用。使用仪器的漫反射模式,样品池为Harrick公司生产的可通气体的原位样品池,使用液氮冷却的Mercury-Cadmium-Telluride(MCT)检测器记录光谱信号。待样品还原后降至室温取背景,然后通入1.4%PO/He并保持30 min使其吸附饱和,随后切换为Ar吹扫40 min并取光谱,二者作差谱获得PODRIFTS结果。
1.3 性能考察
环氧丙烷异构化和深度氧化反应的性能考察在配有在线色谱仪的连续流动固定床装置上进行。具体步骤如下:称取0.15 g堵孔TS-1-B分子筛或Au/TS-1-B催化剂,将过筛后的样品(<110μm)装进内径为6 mm的石英反应管中。采用40%的H2/N2混合气对分子筛或催化剂预处理,流量为50 ml∙min-1,升温至300℃并保持2 h,调节反应器床层温度至200℃并稳定一段时间后,调节气体流量使PO体积分数为0.4%的PO/N2混合气体通过床层进行反应,氢气与氧气的体积分数均为10%,总流量为35 ml∙min-1,质量空速为14000 ml∙h-1∙g-1。采用在线气相色谱仪分析反应器出口物质组成,其中PO、丙醛、丙酮和乙醛采用Restek公司生产的RT-QS-Bond色谱柱进行分离并通过氢离子火焰检测器(FID)进行分析,H2和CO2采用TDX-01色谱柱进行分离并利用热导检测器(TCD)进行分析。各物质的相对校正因子均经过已知浓度的气体进行标定。PO转化率、产物选择性以及碳平衡通过式(1)~式(6)进行计算。

式中,NPO,in为反应器进口中PO分子物质的量,mol;NPO,out、N丙醛,out、N丙酮,out、N乙醛,out、NCO2,out分别为反应器出口中PO、丙醛、丙酮、乙醛和二氧化碳分子物质的量,mol。
1.4 模型构建与计算方法
理论计算在Gaussian 09软件包上进行,TS-1模型以MFI结构的SiO2分子筛为基础截取团簇模型,将T6位点的Si原子替换为Ti原子,构造T5位点缺陷,团簇末端的Si悬断键采用H原子进行饱和,并设置Si—H键的键长为0.146 nm[21-23]。计算中所使用的泛函和基组为B3lyp 6-31G(d,p),并结合D3BJ方法对体系的色散作用进行校正[24-26]。构建三足的Ti-Defect类型结构,即由一个钛羟基与三个硅羟基组成,Wells等[21]研究发现该结构相比于四配位Ti(SiO)4位点具有更高的丙烯环氧化活性。计算中涉及的所有吸附态和过渡态均进行了频率验证,以确保吸附态没有虚频以及过渡态有且仅有一个虚频,并对反应过渡态进行了IRC分析,验证过渡态虚频的振动方向为化学反应进行的方向。计算吸附能和过渡态的能垒采用各状态的电子能量进行计算,并经过零点能和基组重叠误差(BSSE)的校正。限制性优化后的9T分子筛模型结构如图2所示,对该团簇模型中四个Ti—O键的键长进行分析(表1),发现它们介于0.175~0.182 nm之间,其平均值为0.179 nm,这一数值与文献中EXAFS实验所测得的(0.1793±0.0007)nm接近[27]。
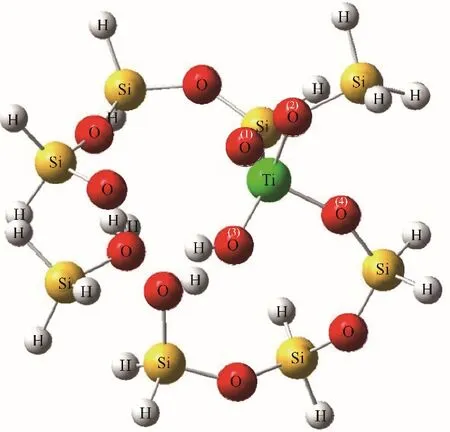
图2 计算中所采用的TS-1分子筛9T模型Fig.2 The optimized 9T clusters model of TS-1 employed for the calculation

表1 TS-1模型中Ti—O键键长与实验值比较Table 1 Comparison of Ti—O bond lengths between calculated and experimental values
2 实验结果与讨论
2.1 样品表征与性能考察
图3(a)TS-1-B的XRD谱图表明其具有典型的MFI骨架结构[28-29]。采用UV-Vis光谱分析样品中Ti的配位环境,图3(b)中位于207 nm处的强吸收峰表明TS-1-B中钛物种主要为四配位状态[13],其中290 nm左右的肩峰归属于孔道内模板剂对钛配位结构的影响[15]。图3(c)HRTEM结果显示所合成的分子筛大小较为均匀,粒径为300~400 nm。采用氮气物理吸附表征来探究所合成分子筛的比表面积,图3(d)显示了100℃干燥的堵孔TS-1-B和550℃焙烧的开孔TS-1-O的吸附等温线,其中TS-1-O展现了典型的I型等温线,表明其具有MFI的微孔结构。而TS-1-B由于微孔被堵塞而具有较少的氮气吸附量。表2中给出了两个样品的BET比表面积和基于t-plot方法的孔容数据,结果显示TS-1-B的BET比表面积约为39 m2∙g-1,这与文献中报道的TS-1-B的数据相一致[13]。
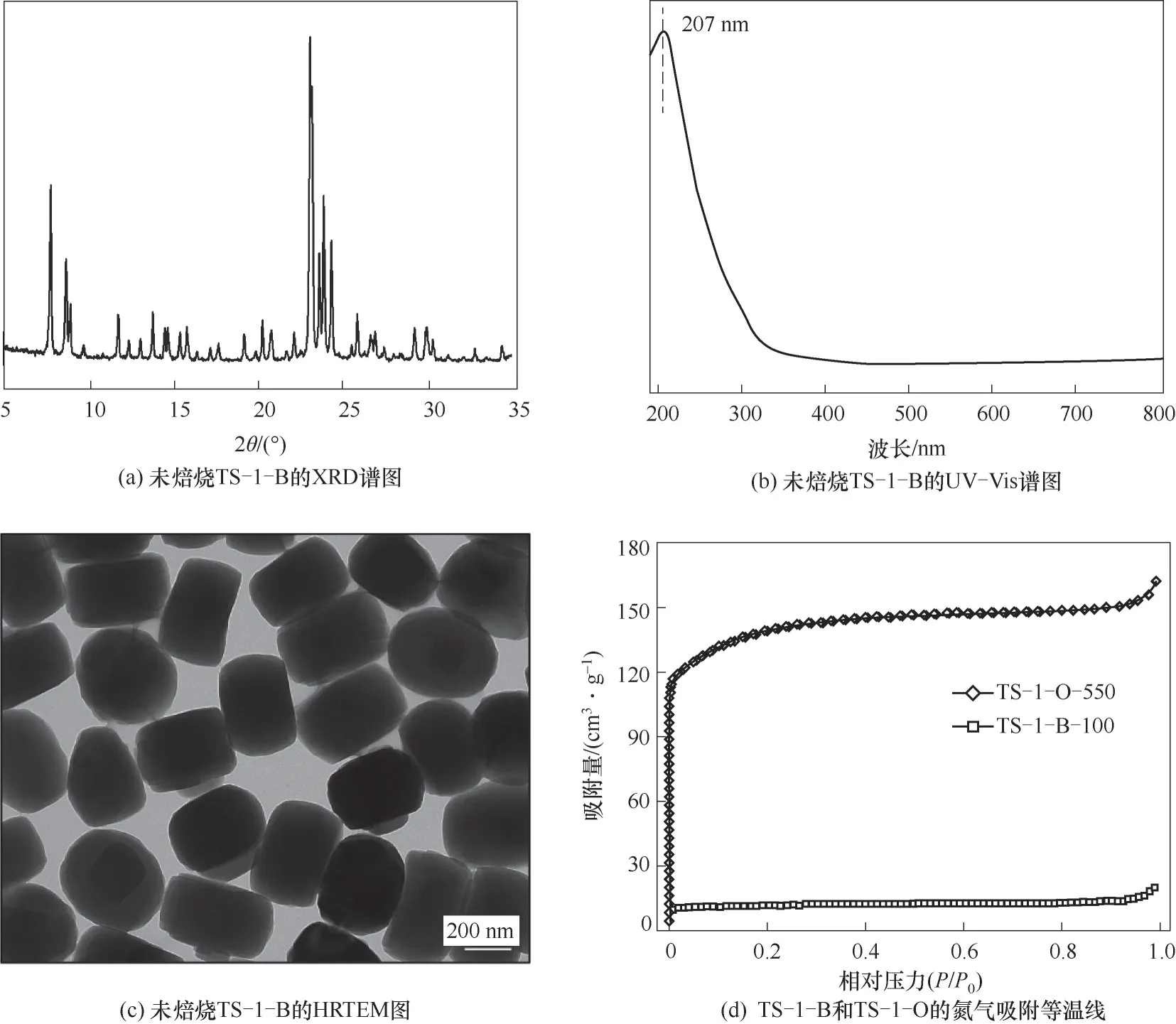
图3 所合成的钛硅分子筛的结构表征Fig.3 Structural characterization of synthesized titanosilicate

表2 TS-1-B和TS-1-O的BET比表面积和孔容参数Table 2 BET surface area and pore parameters of TS-1-B and TS-1-O
通过对还原后Au/TS-1-B催化剂的HAADFSTEM结果中超过200个金颗粒的粒径统计,确定其粒径分布约为(3.1±0.6)nm(图4)。采用FT-IR探究PO在TS-1-B和Au/TS-1-B上的吸附作用,图5(a)PO-DRIFTS结果显示二者具有相似的PO吸附量,表明PO主要吸附在钛硅分子筛而不是纳米金颗粒上。采用不同气氛下的反应结果来探究丙烯氢氧环氧化反应中丙醛和丙酮副产物的生成路径。首先,向Au/TS-1-B催化剂上通入摩尔分数均为10%的丙烯与氧气,发现在200℃的反应温度下产物主要是丙烯醛,未检测到明显的PO、丙醛和丙酮。对该TS-1-B分子筛催化PO异构化的性能进行考察,结果如图5(b)所示,观察到产物中有丙醛和丙酮,且丙醛比丙酮具有更高的选择性,这与该分子筛负载的纳米金催化剂上的反应结果相吻合[15-16]。进一步向Au/TS-1-B催化剂上通入PO的结果如图5(c)所示,可以看到催化剂上PO的转化率略微升高,可能是由于金颗粒与载体之间的电子转移作用促进了PO在分子筛上的吸附。而当通入氢气和氧气后,PO的转化率急剧增加,并生成了乙醛和二氧化碳副产物,这可能主要是由于氢气和氧气在Au位点上反应生成的过氧物种与PO发生深度氧化反应,造成了碳碳键的断裂生成乙醛和二氧化碳。这些结果说明了丙烯氢氧环氧化反应中丙醛和丙酮副产物主要来源于PO在分子筛上的异构化反应。
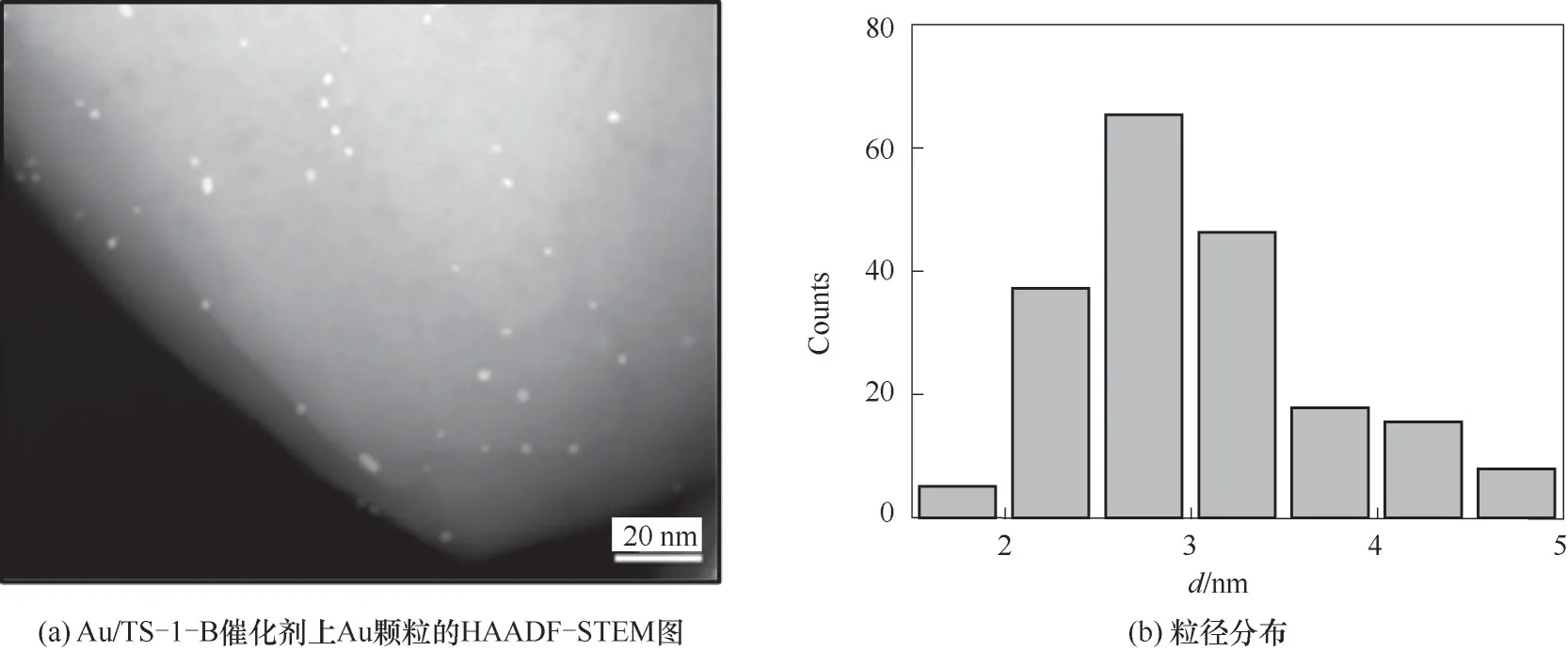
图4 Au/TS-1-B催化剂上Au颗粒的HAADF-STEM和相应的粒径分布Fig.4 Typical HAADF-STEM image and corresponding particle size distribution of Au/TS-1-B catalyst
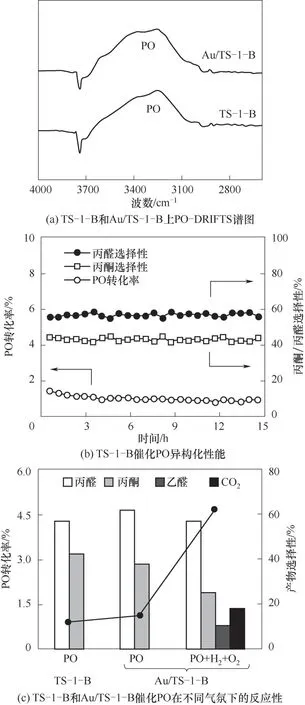
图5 TS-1-B和Au/TS-1-B上PO异构化和深度氧化反应性能Fig.5 Catalytic performance of PO isomerization and overoxidation over TS-1-B and Au/TS-1-B
为了认识PO在TS-1分子筛上的开环和异构机理,采用理论计算探究PO分子在TS-1上转化生成丙醛和丙酮的吸附态以及过渡态。通过对PO分子结构和反应路径的解析可知,PO异构化的反应过程主要有碳氧键断裂和氢原子转移重排两个过渡态[20,30]。由于PO分子中存在两个C—O键,C1—O和C2—O键键长分别为0.143 nm和0.144 nm,断裂后将分别朝向丙酮和丙醛的生成。下文将对这两种情况分别进行计算研究和讨论。
2.2 PO异构生成丙醛反应机理
PO异构生成丙醛过程中所涉及的关键结构和键长参数如图6所示,PO首先吸附在TS-1酸性位点附近,可以看到PO分子主要通过其电负性的氧原子与Ti原子和羟基发生相互作用。通过分析吸附态PO中氧原子与其他原子的距离可知,PO与硅羟基上氢原子的距离(0.244 nm)比其与钛羟基上氢原子的距离(0.279 nm)更近。这说明,相比于钛羟基,PO与硅羟基具有更强的相互作用。动力学研究和红外表征揭示这种吸附作用不仅占据了丙烯环氧化的活性位,对PO生成速率具有抑制效应,同时也会促进PO分子进一步开环异构和深度氧化生成副产物[31-33]。相比于气相中C—O键键长,吸附态的键长得到了一定程度的拉伸,其中C2—O键的键长被拉伸至0.145 nm,预示着PO分子的活化,该吸附过程对于PO中三元环结构的碳氧键断裂具有重要作用[20,34]。与二号碳原子相连的碳氧键在频率振动中受到吸附作用的牵引,形成该化学键断裂的过渡态,如图6(b)所示,其中C2—O键的键长被进一步拉伸至0.215 nm。随后与Ti相连的骨架中的氧原子牵引二号碳原子形成五元环的中间体结构,即双配位丙氧基物种[图6(c)]。大量光谱研究的结果证实了该物种在丙烯氢氧环氧化反应中的存在,其也被广泛认为会进一步衍变成碳酸盐或羧酸盐物种并导致催化剂失活[19,35-38]。进一步,一号碳原子上的氢原子在频率振动中受到二号碳原子的牵引,形成氢原子转移重排的过渡态,同时C1—O键的键长缩短至0.132 nm,如图6(d)所示。随后二号碳原子捕获到该转移的氢原子,一号碳原子与氧原子形成键长为0.122 nm碳氧双键,生成吸附态的产物丙醛。
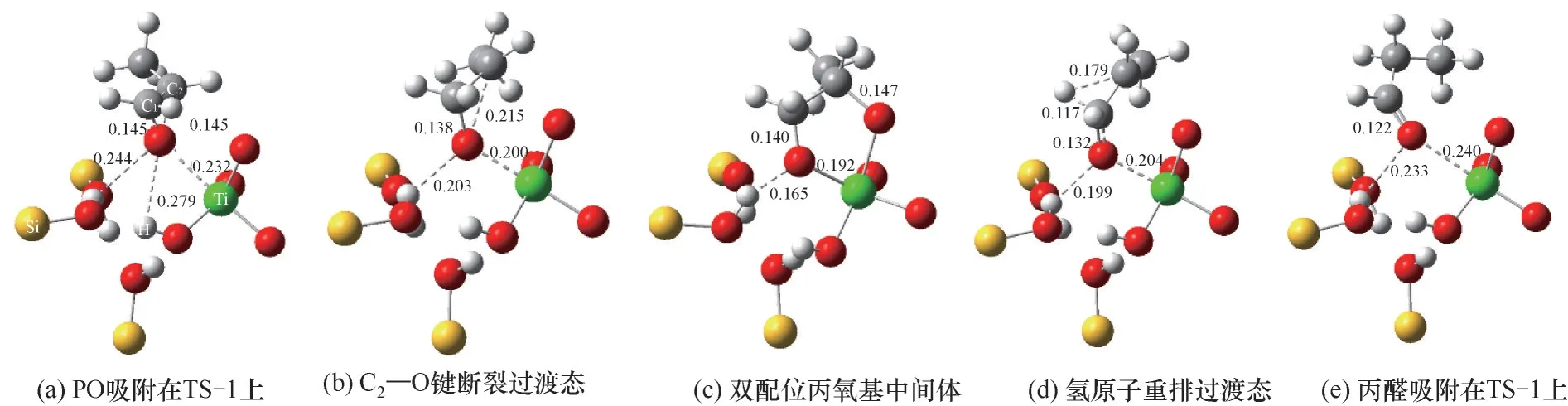
图6 TS-1催化PO异构化生成丙醛中的关键结构和键长(单位:nm)Fig.6 The key structures and bond lengths during PO isomerization to produce propanal over TS-1(Unit:nm)
上述过程的能线图如图7所示,PO在该Ti位点上的吸附能为43 kJ∙mol-1,C2—O键断裂的过渡态与吸附态之间的能垒为152 kJ∙mol-1,而碳上氢原子转移重排的过渡态与吸附态之间的能垒为106 kJ∙mol-1。通过对这两个过渡态的频率进行分析,发现其虚频的振动频率分别为-323 cm-1和-443 cm-1,从气相反应物PO到气相产物丙醛的过程中释放能量约为96 kJ∙mol-1。

图7 TS-1催化PO异构化生成丙醛的能线图Fig.7 Energy profile for PO isomerization to produce propanal over TS-1
2.3 PO异构生成丙酮反应机理
在PO异构形成丙酮过程中,PO在钛位点附近与钛原子、硅羟基以及钛羟基发生吸附作用,并随后经过碳氧键断裂和氢原子转移重排两个过渡态。与PO异构形成丙醛所不同的是,丙酮的形成需要断裂PO分子中一号碳原子上的碳氧键,并与钛原子形成五元环结构的双配位丙氧基物种中间体,随后该物种二号碳原子上的氢原子发生重排向一号碳原子转移,生成吸附态的产物丙酮,各关键步骤的结构和键长参数如图8所示。

图8 TS-1催化PO异构化生成丙酮中的关键结构和键长(单位:nm)Fig.8 The key structures and bond lengths during PO isomerization to produce acetone over TS-1(Unit:nm)
PO异构形成丙酮过程的能线图如图9所示,C1—O键断裂和氢原子重排过程两个过渡态的振动频率分别为-454 cm-1和-435 cm-1,其与吸附态的能垒均比丙醛的生成过程对应的能垒要高,尤其是对于氢原子重排过程,达到了162 kJ∙mol-1。这可能是由于丙酮生成过程中,二号碳原子上的氢向一号碳原子转移时会受到三号碳原子反方向的吸引力,造成了过渡态的能量较高。从气相反应物PO到气相产物丙酮的过程中释放能量约为130 kJ∙mol-1。

图9 TS-1催化PO异构化生成丙酮的能线图Fig.9 Energy profile for PO isomerization to produce acetone over TS-1
通过把PO异构的两条路径整个过程进行对比可以看到,丙醛生成过程的能垒(152 kJ∙mol-1)小于丙酮生成过程的能垒(162 kJ∙mol-1),预示着丙醛相比于丙酮更容易生成。这一结果与前文中PO异构化结果以及文献中Au/TS-1-B催化剂上丙醛的选择性比丙酮高相一致[15-16],也与文献报道中ZSM-5分子筛上PO异构化的理论计算结果相吻合[20]。
基于以上研究结果,PO在钛硅分子筛表面与钛原子和羟基具有较强的相互作用,这不仅会引起PO的开环反应转化形成副产物,也可能会占据丙烯环氧化的活性位,从而抑制PO活性。目前已有相关的研究工作报道采用硅烷偶联剂提高催化剂的疏水性来减弱PO吸附从而增强丙烯环氧化性能[39-41]。本文的研究结果将为钛基催化剂的结构改性以增强PO脱附从而调控PO活性和选择性提供理论依据。
3 结 论
本文基于TS-1-B分子筛和Au/TS-1-B催化剂上不同气氛下的FT-IR和性能考察结果,发现丙烯氢氧环氧化反应中丙醛和丙酮的生成主要来源于PO在钛硅分子筛上的异构化反应。进一步对TS-1催化PO异构反应的机理进行了研究,采用DFT计算方法优化了TS-1上Si缺陷的Ti-Defect位点结构,探究了PO异构生成丙醛和丙酮的反应路径及其各状态对应的能量。结果显示PO异构化需经历碳氧键断裂、形成五元环结构双配位丙氧基中间体和碳上氢原子转移重排等关键步骤。其中,相比于丙醛,丙酮的生成过程中由于C2上氢原子向C1转移重排的过渡态能垒较高导致了丙酮在PO异构化反应中更难生成。
