群体生物等效性用于吸入制剂的探讨
2021-10-14陈立营徐景娜周学海
陈立营,徐景娜,杨 敏,周学海*
(1.天津药业研究院股份有限公司,天津300386;2.天津市医药集团技术发展有限公司,天津300300)
吸入制剂为局部作用的药物,药物首先被递送到肺部,而后进入体循环,同时还通过其他部位如口、咽、胃肠道等进入体循环,药代动力学和局部递药等效性之间关系复杂,通常仅采用药代动力学方法不足以评价吸入制剂的生物等效性[1]。对于吸入制剂,FDA建议使用证据权重法进行吸入制剂产品的等效性研究,吸入制剂必须同时满足处方及给药装置相似、体外测试等效、药动学研究(pharmacokinetics,PK)等效以及药效学(pharmacodynamics,PD)和临床研究等效,生物等效性才能成立[2]。欧洲药品管理局(EMA)指南推荐使用逐步评估的方法进行吸入制剂的生物等效性研究,该方法认为只需满足体外等效性即可,若体外不满足,则需结合体内的方法判断其生物等效性[3]。生物等效性分为平均生物等效性(average bioequivalence,ABE)、群体生物等效性(population bioequiva lence,PBE)和个体生物等效性(individual bioequiva lence,IBE)。目前应用较多的是ABE,ABE只要求参比制剂与受试制剂在平均水平等效,PBE不仅要求参比制剂与受试制剂在平均水平等效,也要求参比制剂与受试制剂在变异方面等效。与ABE相比,PBE更关注制剂的多变性[4]。
吸入制剂为药械组合产品且为局部给药,由于剂型的复杂性,受药学、装置、患者使用情况及环境等的影响,变异性较大,FDA建议吸入制剂需先采用PBE方法进行体外研究,再结合药动学指标和临床终点指标共同评价吸入制剂仿制制剂的安全性和有效性[5]。之前EMA和国家食品药品监督管理局(NMPA)认为运用ABE评价方法即可以满足要求,对PBE和IBE方法经验有限。在2020年12月NMPA公布了《群体药代动力学研究技术指导原则》,NMPA也开始重视PBE。本文根据FDA相关指导原则剂及相关文献,对吸入制剂的PBE统计分析方法、体外评价参数等进行了介绍,并对影响PBE结果的因素进行了评价,为吸入制剂体外生物等效性研究提供思路。
1 吸入制剂的PBE研究参数及要求
吸入制剂需要进行体外研究的参数有单喷含量、空气动力学粒径分布、递送剂量均一性、喷雾模式、喷雾几何、首次灌注、再次灌注、持续时间、喷雾速度等。其中空气动力学粒径分布的ISM(impactor sized mass)用于PBE研究,质量中值粒径、微细粒子量、微细粒子分数、几何均值及各级分布数据作为支持性数据进行提交。喷雾模式需测定两个距离下(3~7 cm)的椭圆率和面积。FDA要求进行PBE研究参比制剂与受试制剂至少需要各3批,每批各10支,每支需进行不同生命周期的考察,即生命前期、后期、中期。此外,吸入制剂分为定量吸入气雾剂、吸入粉雾剂、吸入软雾剂等,吸入制剂的不同剂型、不同参数进行PBE研究的要求不同,吸入制剂进行PBE研究的参数及要求见表1。
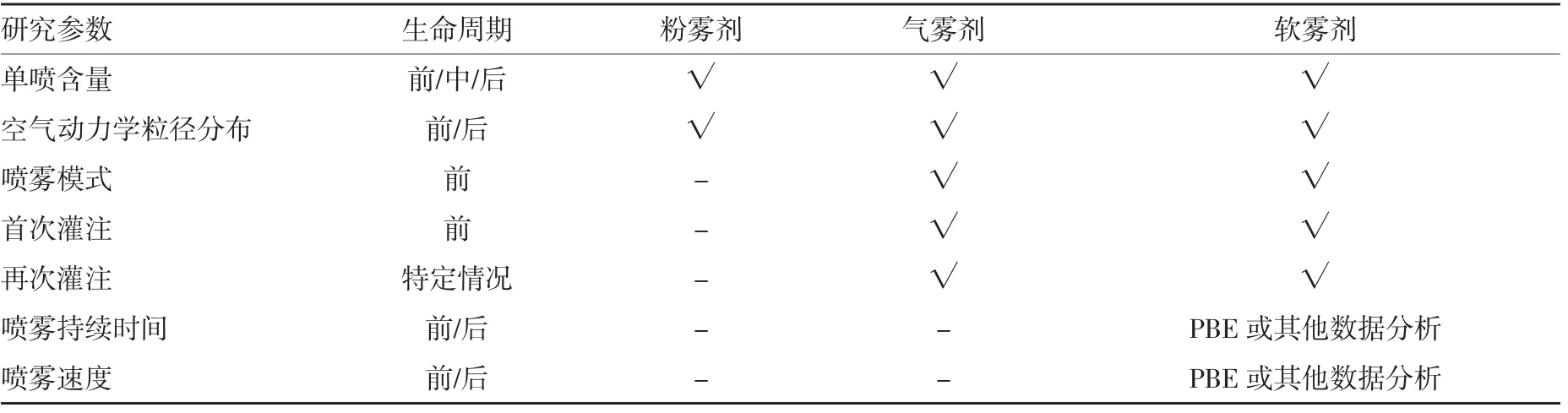
表1 吸入制剂PBE研究的参数
2 PBE统计分析原理
PBE概念是在1992首次提出,1997年写入FDA指导原则[6],经过数次更新于2012写入布地奈德BE草案中,此后吸入制剂更新的BE指南中关于PBE的方法均以此作为参考。计算见公式1-2[7]。
公式1:[(μT-μR)2+(σT2-σR2)]/max(σT02,σR2)≤θp
公式2:θp=(ABElimit+variance termoffset)/Scaling variance=[(ln1.11)2+εp]/σT02
由公式1、2知,PBE在ABE的基础上,增加了变异的情况,PBE更关注制剂的多变性。公式中,(μT-μR)为受试制剂与参比制剂的均值(对数)差,σT2、σR2为受试制剂与参比制剂的总方差,σT0、εp为法定常数。FDA认为,与体内生物等效性相比,体外生物等效性变异更小,公式中ABE上限值取ln1.11,εp取0或0.01,布地奈德BE草案文件中取值为0.01,σT0取值为0.1,经计算θp为2.089[8]。
对于评价标准,FDA推荐使用混合标度法,避免参比变异过大,造成假阳性的结果。
对于σT0>σR,采用常数标准,η1=(μT-μR)2+(σT2-σR2)-θpσT02<0。对于于σT0<σR,采用参比标准,η2=(μT-μR)2+(σT2-σR2)-θpσR2<0。进行评价时,相应标准下η1或η2的95%置信区间的上限值Hη≤0,则可认为受试制剂与参比制剂的PBE等效。
3 PBE实例应用
3.1 PBE结果影响因素考查 根据FDA布地奈德BE草案文件中PBE的计算方法,用SAS软件进行编程,并运行布地奈德BE草案的参比与受试数据,验证软件及程序的可行性。BE草案中,σR值为0.404 6,Hη值为-0.031 698 721,重现结果σR值为0.404 6,Hη值为-0.031 498 674。重现结果与BE草案中的结果基本一致,认为软件及程序可行。
BE草案中,参比制剂与受试制剂数据均为3批,每批10支,每支进行了不同生命周期(前中后)的研究,其中参比制剂三批总均值为5.858 151,RSD值为6.83%。利用EXCEL随机数据发生器,参考BE草案参比制剂数据范围,随机生成数据,选择一组均值与参比制剂相近,但RSD值与参比制剂有一定差异的数据作为模拟1,并作为受试制剂数据,以BE草案中的参比数据作为参比数据,运行PBE,运行结果见表2。利用EXCEL随机数据发生器,参考BE草案参比制剂数据范围,随机生成数据,选择一组RSD与草案参比制剂相近,但均值与参比制剂有一定差异的数据作为模拟2,并作为受试制剂数据,以BE草案中的参比数据作为参比数据,运行PBE,运行结果见表2。并将运行PBE的草案参比数据、草案受试数据、模拟1数据及模拟2数据进行作图比较,见图1。
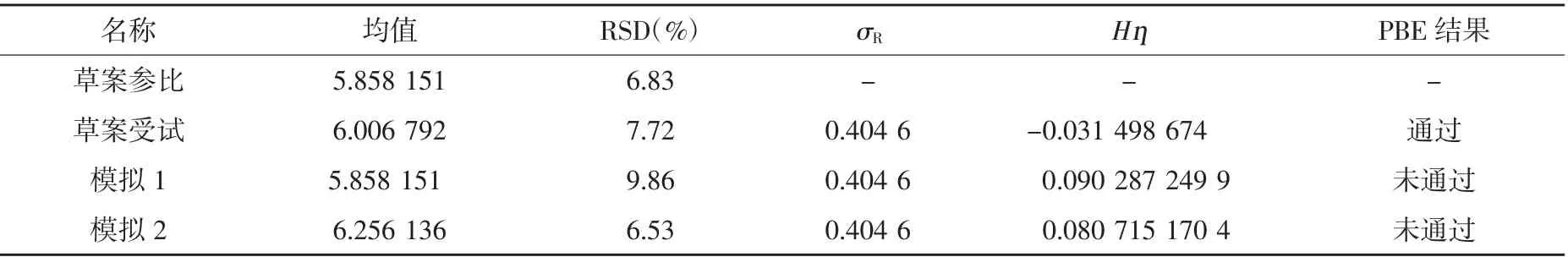
表2 PBE模拟运行结果表

图1 PBE数据比较图
模拟1中,均值与参比一致,参比RSD值为6.83%,受试RSD值为9.86%,差异较大,PBE模型未通过。模拟2中,RSD值与参比一致,参比三批总体均值为5.858 151,受试三批总体均值为6.256 131,总体均值差异较大,PBE模型未通过。由图1可知,模拟1受试数据与参比数据相比,分布较离散,批内变异大,导致PBE未通过。模拟2受试数据与参比数据相比,模拟值主要分布在参比数据上边缘值附近,总体均值差异较大,导致PBE未通过。因此,在PBE模型评价中,影响PBE结果的因素有数据的均值、变异及分布等。受试制剂需在均值、变异及分布上均要与参比制剂相近才能保证PBE等效。
3.2 PBE参数研究考查 根据PBE模型对个各参数的要求,原研制剂与自研制剂各准备了3批,每批10支,对单喷含量(SAC)、空气动力学粒径分布下的撞击微粒子量(ISM)、喷雾模式下的椭圆率(Ovality)和面积(Aera)进行了研究,并运行PBE模型,各参数运行结果见表3。参比制剂SAC、ISM、Aera的总方差(σR)均大于0.1,变异性较大,为参数标准。Ovality的总方差(σR)均小于0.1,变异性较小为常数标准。这4个参数中只有Ovality通过PBE模型。相同受试制剂不同参数通过PBE的可能性不同,为提高BE通过的可能性及用药安全性,需保证各参数均通过PBE模型。根据各参数通过PBE的情况,进而指导处方工艺研究。
由表3可知,参比制剂SAC、ISM、Aera的总方差(σR)均大于0.1,变异性较大,为参数标准。Ovality的总方差(σR)均小于0.1,变异性较小为常数标准。这4个参数中只有Ovality通过PBE模型。相同受试制剂不同参数通过PBE的可能性不同,为提高BE通过的可能性及用药安全性,需保证各参数均通过PBE模型。根据各参数通过PBE的情况,进而指导处方工艺研究。
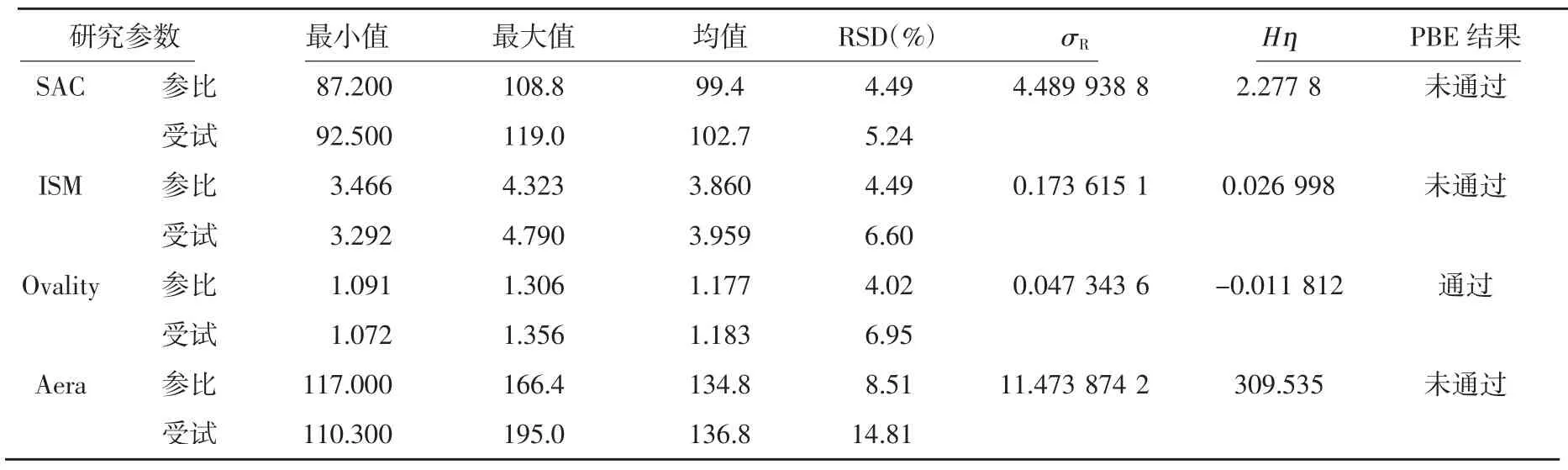
表3 各参数PBE运行结果
4 讨论
吸入制剂的药械组合及局部给药的特殊性,均决定了其对体外生物等效性的要求更高,不同药监局对吸入制剂的生物等效性要求不一样。随着各国对吸入制剂的研究,越来越多的体内外模型用于吸入制剂的生物等效性研究。例如美国的FDA用PBE模型来评价吸入制剂的体外等效性。PBE要求吸入制剂的受试制剂要在均值、变异及分布与原研制剂保持一致,进而保证批间及批内差异一致,保证患者的用药安全性。在进行处方工艺研究时,相同受试制剂各参数通过PBE的可能性不一样,在进行处方工艺与装置研究时,应保证各参数均能通过PBE。根据PBE的运行结果,及时调整处方工艺,提高研发效率,降低研发成本。此外,还可以通过PBE去评价各参数的变异性,对各参数在吸入制剂性能评价中的权重进行分析,进而指导研究方向,这也是后期需进一步关注的地方。
